ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2014; 2:89-93. doi:10.7150/jgen.7298 This volume Cite
Research Paper
Enly: Improving Draft Genomes through Reads Recycling
Marco Fondi1 ![]() , Valerio Orlandini1, Giorgio Corti2, Marco Severgnini2, Marco Galardini1, Alessandro Pietrelli2, Fabio Fuligni2, Michele Iacono2, Ermanno Rizzi2, Gianluca De Bellis2, Renato Fani1
, Valerio Orlandini1, Giorgio Corti2, Marco Severgnini2, Marco Galardini1, Alessandro Pietrelli2, Fabio Fuligni2, Michele Iacono2, Ermanno Rizzi2, Gianluca De Bellis2, Renato Fani1
1. Dept. of Evolutionary Biology, Via Madonna del Piano 6, 50143 Sesto Fiorentino,Florence, Italy;
2. Istituto di Tecnologie Biomediche, Consiglio Nazionale delle Ricerche (ITB-CNR), Segrate (MI), Italy.
Published 2014-4-5
Abstract
The reconstruction of the complete genome sequence of an organism is an important point for comparative, functional and evolutionary genomics. Nevertheless, overcoming the problems encountered while completing the sequence of an entire genome can still be demanding in terms of time and resources. We have developed Enly, a simple tool based on the iterative mapping of sequence reads at contig edges, capable to extend the genomic contigs deriving from high-throughput sequencing, especially those deriving by Newbler-like assemblies. Testing it on a set of de novo draft genomes led to the closure of up to 20% of the gaps originally present. Enly is cross-platform and most of the steps of its pipeline are parallelizable, making easy and fast to improve a draft genome resulting from a de novo assembly.
Keywords: Genome assembly, Next Generation Sequencing, Contig extension, Newbler, 454.
Introduction
Sequence assembly is the first challenge encountered in a typical computational genomics pipeline and it involves the merging and the ordering of shorter sequence fragments (reads) with the aim to get as close as possible to the original larger sequence (genome). Advent of next-generation sequencing platforms has allowed the sequencing a huge number of organisms and species at reasonable costs. However, many issues regarding the computational assembly of large-scale sequencing data have remained unsolved [1] and, actually, the number of draft genomes in databases greatly overtakes the number of completely sequenced (sometimes referred also as “finished” or “closed”) ones (www.genomesonline.org). The output of a de novo assembly is typically a draft genome, consisting of a set of contigs (i.e. contiguous sequence fragments) that may be ordered and oriented into scaffold sequences, with gaps between them, representing regions of uncertainty or missing sequence [2]. Alternatively, draft genomes can be represented in the form of (De Bruijn) graphs, an approach that is currently exploited by a number of assembly algorithms (reviewed in [3]). The difficulty in obtaining a closed genome may be due to several causes, including the presence of repetitive fragments along the genome and/or the absence of enough reads to produce a reliable assembly. It is also known that the most rapidly evolving regions are often absent or incorrectly rendered in finished genomes [4]. Furthermore, from a mathematical viewpoint, the de novo genome assembly problem can be proven to be difficult, falling within a class of problems (NP-hard) for which no efficient computational solution is yet known [5]. In this context, it must be added that some traditional assemblers like Newbler (454 Sequencing, Roche Diagnostics, Indianapolis, IN, USA) usually perform a trimming of the contig edges depending on the quality of the supporting reads. This conservative procedure, however, may result in a loss of information, discarding many correct bases characterized by a sub-optimal quality. To overcome these limitations, we have developed a tool called Enly that allows increasing the length of the contigs deriving from de novo assemblies and the closure of part of the gaps commonly present in the draft genome. Enly is sequencing platform-independent since it be used with any kind of sequence type, as long as files are provided in Fasta format. Accordingly, even hybrid datasets (obtained with different sequencing technologies) can be used as input for Enly pipeline. Finally, by taking into account scaffold information, Enly drastically reduces the probability of generating chimeric assemblies.
Results and Discussion
The overall procedure requires (at least) two multi-Fasta files as input, one containing all the contigs deriving from the first assembly and the other all the raw reads resulting from the sequencing run. By applying the strategy described below to each contig in the input file, Enly tries to increase their length and (possibly) to merge them into scaffolds.
More in detail, the pipeline is based on the iteration of multiple cycles (Figure 1) and during each of them:
Schematic representation of the whole Enly pipeline. The pipeline starts taking the multi-fasta file embedding all the contigs as input and, for each contig, the following extension procedure is performed. 1) A fragment of a given number of bases is detached from one of the two extremities and is used as query against the reads database (default fragment length is 500 bp but the user can specify this value with the -i option). 2) The BLAST output file is parsed and only those protruding reads partially aligned at the end of the contig and protruding from its extremity are maintained; 3) these reads and the original contig are assembled together to obtain a possibly enlarged contig; 4) steps 3 and 4 are repeated for the other end of each contig. After all the contigs have been probed, the first step ends and 5) the second begins. During the second step, a shorter fragment is detached (default value is 50 bp shorter than the previous step but the user can specify this parameter with the -d option). The procedure ends when the minimum fragment length is reached (option -m) and, at this point, if a reference genome has been provided (-r option) 6) enlarged contigs are mapped onto the reference genome. In phase 7) overlaps among extended contigs are checked against a reference scaffold generated during step 6) or against a reference scaffold provided by the user (-y option) and, eventually, 8) contigs are merged and the contigs and scaffold files are updated. At this point a new cycle begins (unless the maximum number of cycles specified by the user with the -c option has been reached and/or no bases have been added to the contigs during the last cycle). Note that green arrows indicate steps performed only if -r or -y options are selected by the user and that red arrows represent the iteration of the different steps within the same cycle. As an example, setting the -b parameter to 500, -m to 200 and -d to 50 will cause Enly to probe, at every cycle, the reads database with contigs extremities of length 500, 450, 400, 350, 300, 250 and 200 bp.
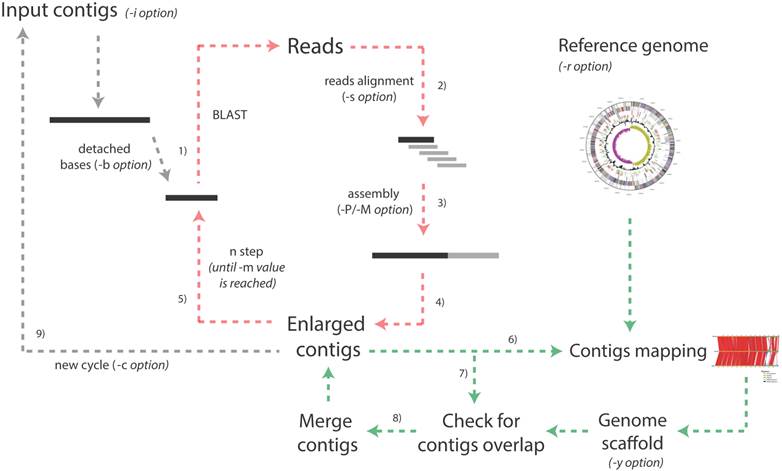
1) A fragment of length l1 (specified by the user) is detached from the 5'-end of each contig and is used as an input for a BLAST search [6] against a database embedding all the original reads resulting from the sequencing run;
2) The BLAST output is parsed to identify reads that can be used to extend the contig (i.e. those partially aligned at the end of the contig and protruding from its extremity).
By default identity threshold among the reads and the contig's extremity is set to 97%. Overlap length between each read and the contig extremity is, instead, specified by the user (with the -s parameter). This means that, for example setting -s to 70%, only reads aligned for at least 70% of their length and sharing more than 97% of their sequence with the contig extremity will be used to extend contigs. Intuitively, setting this overlapping threshold to higher values will increase accuracy, although reducing the number of closed gaps (see Additional file 1: Supplementary Material). The use of higher thresholds (i.e. higher stringency) is, therefore, recommended when a reference genome/scaffold is not available for checking possible chimeric joints (see point 8). When a reference scaffold is present, contigs merging will still be reliable even when low overlap thresholds (e.g. 30%) are selected, although it must be pointed out that the extended contigs not merged into any (sub) scaffold would not be checked for the presence of wrongly incorporated sequence.
3) The selected reads and the original contig are assembled together using either Phrap [7] or Minimo [8] assemblers, possibly resulting in an “enlarged” contig (i.e. contig of increased length).
4) The very same procedure is repeated for the 3'-end of the contig and for all the other contigs of the input file;
5) The extended contigs are used as inputs for a second step, in which a fragment of length l2 (with l2< l1) is detached from each contig end and used as an input for a further BLAST against the reads database, assembling the matching reads with the enlarged contig. This procedure, performed in order to compensate for the presence of reads with a heterogeneous length distribution (resulting from a typical 454 run [9]), is repeated for fragments of decreasing length, until the minimum length (specified by the user through -m option) has been reached;
6) If a reference genome (in Fasta format) has been provided by the user, extended contigs are mapped onto it, taking advantage of an ad hoc modified version of the CONTIGuator tool [10], allowing launching this tool in an iterative fashion and storing results from each run in a separate folder.
7) Contigs are mapped by BLAST one against each other for identifying possible overlaps and gaps closure. No threshold for contigs overlap is set in this stage since scaffold information is used (see next point) to discard possible chimeric joints.
8) Gap closures are validated against a reference scaffold, provided by the user (-y option) or, alternatively, generated by the CONTIGuator tool. Only contigs overlaps that are consistent with scaffold information are merged (using the “megamerger” tool from the EMBOSS suite). In case neither a scaffold nor a reference genome have not been provided by the user, possible overlaps for each of the Enly cycles are stored in a specific output files (see Enly's manual for details) that the user can manually inspect for eventual gap closures.
9) These contigs, together with those that have not been enlarged/merged are then used as input for the following cycle of the pipeline.
The first cycle of the Enly pipeline is completed when all the contigs have been processed, mapped and saved to a new multi-Fasta file. The procedure is then repeated (re-starting from point 1) for a user-specified number of cycles or, alternatively, until no more bases have been added to the contigs during the last cycle. Output files from the mapping procedure (CONTIGuator) are saved in separate folders (one per cycle), ready for being loaded by the Artemis Comparison Tool [11], in order to visually inspect contigs alignment against the reference genome. Reads used for extending contigs may derive from any of the currently available sequencing technologies (Illumina, 454, IonTorrent, etc.), as long as they are provided in the form of Fasta files. Moreover, reads obtained by different sequencing technologies may be pooled in a single file to be used as input for Enly pipeline. Intuitively, since Enly uses the sequence of the reads partially protruding from each contig, the probability of extending its length increases with the average length of the reads dataset. It should be noticed that, besides 454 pyrosequencing, other sequencing platforms are now starting to generate reads of length comparable to the ones of the datasets used in this work (e.g. Illumina MiSeq, IonTorrent), thus paving to the use of this alternative datasets for gap-filling through Enly's approach.
To evaluate the reliability of the pipeline, we tested Enly on three different 454 reads datasets retrieved from either the NCBI short read archive database (SRA, http://www.ncbi.nlm.nih.gov/sra), namely Escherichia coli KO11 (SRS084754) and Staphylococcus aureus 649 (SRS114535), or from a previous sequencing run on Streptococcus pneumoniae AP200 [12]. For these datasets the corresponding complete genomes (E. coli KO11 and S. pneumoniae AP200 [12, 13]) or the genome of a phylogenetically close bacterium (S. aureus COL [14]) were available, allowing the validation of the results obtained with Enly. Each reads dataset was first assembled with Newbler v. 2.6, using default parameters, resulting in 719, 254 and 124 contigs for E. coli KO11, S. aureus 649 and S. pneumoniae AP200, respectively. Contigs obtained from de novo assembly were, then, used, together with the corresponding reads, as input for the Enly pipeline. Parameter values (and an evaluation of their effects on the genome assembly) used during these tests are reported in Additional file 1: Supplementary Figure 1, together with introduced mismatch rates for different reads alignment thresholds (-s option, Additional file 1: Supplementary Figure 2 and 3).
In all cases, Enly was able to improve the de novo assembly (Table 1). In detail, the number of closed gaps ranged from 16% to 19.1% of all the gaps present in each input draft genome. Most of the closed gaps resulted from merging two contigs although, in some cases (e.g. E. coli KO11 drafts assembly), up to 5 different contigs were merged in a single (sub) scaffold. Importantly, after running Enly on the de novo assemblies, an increase in the N50 value for each of the genome was observed, accounting for the overall improvement of the input draft assemblies. Interestingly, Enly showed to perform reasonably good also when non-454 reads were used as input for the pipeline. Results obtained with reads obtained from IonTorrent, MiSeq and PacBio sequencing runs are reported in Additional file 1: Supplementary Table 2 and 3.
Enly results on three different reads dataset. Enly's performances on three different 454 reads datasets, E. coli KO11, S. aureus 649 and S. pneumoniae AP200. Percentages in parentheses indicate the relative number of closed gaps in respect to the ones originally present in the draft genome. Enly v1.2 (see the manual of the program for resolving all the dependencies of the pipeline) was used to produce the results shown here. Options used are reported in Additional file 1: Supplementary Material. * Genome size of S. aureus COL [14].
| Strain name | Genome size (Mb) | N. reads | Av. reads length | N.contigs before Enly | Closed gaps | N50 before/after Enly (% variation) |
|---|---|---|---|---|---|---|
| KO11 | 4.92 | 339,220 | 215.7 | 719 | 115 (16%) | 13737/14906 (+8.5%) |
| 649 | 2.80 | 122,569 | 245.6 | 254 | 47 (19.1%) | 22831/27447 (+20.2%) |
| AP200 | 2.13* | 152,452 | 231.2 | 124 | 23 (18.5%) | 40646/53040 (+30.5%) |
The computational time required by Enly correlates with the amount of contigs/reads embedded in the input files and with the number of cycles required by the user. On a machine with eight 3.1 GHz processors, the computational time required for the tests performed in this work (and according to the parameters specified in Additional file 1: Supplementary Table 1) ranged from 2 up to 5 hours depending on the size of the reads datasets and parameters used (see Additional file 1: Supplementary Figure 4).
Recently, two scaffolding approaches apparently similar to the one presented here have been developed. Both of them take advantage of the additional information that is present on a typical Illumina paired-end sequencing run [15, 16]. Currently, Enly does not use paired-end information, being able to process single end sequencing reads (as long as they are provided in Fasta format) for contigs extension; moreover, by implementing the possibility to guide contigs extension/merging through the use of scaffold information (independently generated by the user or obtained through the implemented CONTIGuator tool), our pipeline reduces the probability of chimeric scaffolds.
In conclusion, Enly is a simple, cross-platform and parallelizable tool allowing the improvement of draft genomes resulting from de novo-assembled high-throughput sequencing reads. It is based on the iterative mapping of reads at contigs ends and it is also able to use (and generate) scaffold information to guide contigs merging, reducing the probability of chimeric scaffolding.
Testing it on a set of de novo draft genomes led to the closure of up to 20% of the gaps originally present, thus resulting particularly helpful during genome finishing procedures.
Supplementary Material
Additional File 1Supplementary tables and figures
Acknowledgements
MF is financially supported by a FEMS postdoctoral fellowship (FAF2012). ER is supported by FIRB-Futuro in Ricerca 2008 grant N°RBFR08U07M. We also thank the EU KBBE Project PharmaSea 2012-2016, Grant Agreement no: 312184.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Scheibye-Alsing K, Hoffmann S, Frankel A, Jensen P, Stadler PF, Mang Y. et al. Sequence assembly. Comput Biol Chem. 2009;33:121-36 doi:10.1016/j.compbiolchem.2008.11.003
2. Earl D, Bradnam K, St John J, Darling A, Lin D, Fass J. et al. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome research. 2011;21:2224-41 doi:10.1101/gr.126599.111
3. Compeau PE, Pevzner PA, Tesler G. How to apply de Bruijn graphs to genome assembly. Nature biotechnology. 2011;29:987-91 doi:10.1038/nbt.2023
4. Ribeiro FJ, Przybylski D, Yin S, Sharpe T, Gnerre S, Abouelleil A. et al. Finished bacterial genomes from shotgun sequence data. Genome Res. 2012;22:2270-7 doi:10.1101/gr.141515.112
5. Pop M. Genome assembly reborn: recent computational challenges. Briefings in bioinformatics. 2009;10:354-66 doi:10.1093/bib/bbp026
6. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389-402
7. Machado M, Magalhaes WC, Sene A, Araujo B, Faria-Campos AC, Chanock SJ. et al. Phred-Phrap package to analyses tools: a pipeline to facilitate population genetics re-sequencing studies. Investigative genetics. 2011;2:3. doi:10.1186/2041-2223-2-3
8. Treangen TJ, Sommer DD, Angly FE, Koren S, Pop M. Next generation sequence assembly with AMOS. Curr Protoc Bioinformatics. 2011 Chapter 11: Unit 11 8. doi:10.1002/0471250953.bi1108s33
9. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376-80 doi:10.1038/nature03959
10. Galardini M, Biondi EG, Bazzicalupo M, Mengoni A. CONTIGuator: a bacterial genomes finishing tool for structural insights on draft genomes. Source Code Biol Med. 2011;6:11. doi:10.1186/1751-0473-6-11
11. Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422-3 doi:10.1093/bioinformatics/bti553
12. Camilli R, Bonnal RJ, Del Grosso M, Iacono M, Corti G, Rizzi E. et al. Complete genome sequence of a serotype 11A, ST62 Streptococcus pneumoniae invasive isolate. BMC Microbiol. 2011;11:25. doi:10.1186/1471-2180-11-25
13. Turner PC, Yomano LP, Jarboe LR, York SW, Baggett CL, Moritz BE. et al. Optical mapping and sequencing of the Escherichia coli KO11 genome reveal extensive chromosomal rearrangements, and multiple tandem copies of the Zymomonas mobilis pdc and adhB genes. J Ind Microbiol Biotechnol. 2012;39:629-39 doi:10.1007/s10295-011-1052-2
14. Gill SR, Fouts DE, Archer GL, Mongodin EF, Deboy RT, Ravel J. et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol. 2005;187:2426-38 doi:10.1128/JB.187.7.2426-2438.2005
15. Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13:R56. doi:10.1186/gb-2012-13-6-r56
16. Tsai IJ, Otto TD, Berriman M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010;11:R41. doi:10.1186/gb-2010-11-4-r41
Author contact
![]() Corresponding author: Email: marco.fondiit.
Corresponding author: Email: marco.fondiit.