ISSN: 1839-9940
- Volume 14; 2026
- Volume 13; 2025
- Volume 12; 2024
- Volume 11; 2023
- Volume 10; 2022
- Archive
- Editorial Board
- Cover Images
- Cover Suggestion
- Special Issues
Introduction
Why chromosome dosage matters
Monosomy of the X and the...
Mechanisms of sex-specific X...
Common themes: Recruitment of...
Common themes: Dosage...
Common themes: Transcriptional...
Fine-tuning transcription by the...
Dosage Compensation and...
Conclusion and Perspective
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactJ Genomics 2015; 3:1-19. doi:10.7150/jgen.10404 This volume Cite
Review
Mechanisms of X Chromosome Dosage Compensation
Sevinç Ercan ![]()
Department of Biology, Center for Genomics and Systems Biology, New York University, New York, NY 10003, USA.
Published 2015-1-1
Abstract

In many animals, males have one X and females have two X chromosomes. The difference in X chromosome dosage between the two sexes is compensated by mechanisms that regulate X chromosome transcription. Recent advances in genomic techniques have provided new insights into the molecular mechanisms of X chromosome dosage compensation. In this review, I summarize our current understanding of dosage imbalance in general, and then review the molecular mechanisms of X chromosome dosage compensation with an emphasis on the parallels and differences between the three well-studied model systems, M. musculus, D. melanogaster and C. elegans.
Keywords: X chromosomes, mechanisms
Introduction
Maintenance of correct chromosome dosage is important for development and fitness in many species. Although X chromosome harbors many genes that are important for both sexes, males contain a single X and females contain two X chromosomes. To compensate for X chromosome dosage difference between sexes, different mechanisms have evolved to equalize X-linked transcript levels in males and females. Recent genomic research into the mechanisms of X chromosome dosage compensation in three model organisms, M. musculus (mouse), D. melanogaster (fly) and C. elegans (worm) suggest that although these animals use different strategies, there are considerable parallels in the molecular mechanisms that accomplish X chromosome dosage compensation. Below, first I discuss chromosome and gene dosage in general, and then review different mechanisms of dosage compensation while highlighting the similarities and differences between the molecular mechanisms that regulate X chromosome transcription in mouse, fly and worm.
Why chromosome dosage matters
'Dosage' of a chromosome (or a gene) refers to its genomic copy number. Polyploidies refer to an increase in the dosage of all chromosomes, and are well tolerated. Aneuploidies refer to a change in the dosage of one chromosome with respect to the rest of the genome, and are generally detrimental to the organism (reviewed in [1] and [2]). Partial aneuploidies, due to duplication or deletion of a chromosomal segment, can also be harmful. In some cases, changes in the copy number of a single gene cause problems (e.g. haploinsufficiency). Thus, maintenance of correct gene dosage at multiple scales (single genes to whole chromosomes) is important for an organism's fitness.
Effect of single gene dosage
In general, mRNA and protein levels are directly proportional to gene dosage, as observed in yeast [3, 4], arabidopsis [5], flies [6], mouse [7-9] and humans [10-12]. In fact, altering a gene's copy number is one mechanism of gene regulation. For example in bacteria, antibiotic treatment leads to amplification of genes that increase competence for acquisition of antibiotic resistance genes [13]. In yeast, amplification of genes that are important for nitrogen transport occurs under the selection of nitrogen starvation [14]. In multicellular organisms such as flies, gene amplification in specialized tissues is part of normal development (reviewed in [15]).
Deviations from normal gene dosage can negatively affect an organism's fitness. A classic example is the effect of increased beta-tubulin gene dosage on microtubule formation in yeast [16]. Correct dosage of the transcription factor Bicoid is important for proper body patterning and development in flies [17]. In humans, changes in individual gene dosage have been associated with many diseases (reviewed in [18, 19]). For instance, duplication of vasoactive intestinal peptide receptor gene VIPR2 is associated with schizophrenia [20]; increased copy number of beta-defensin antimicrobial peptides correlates with risk for psoriasis [21]; and copy number variation of the MIR17HG gene (a micro RNAi cluster) is implicated in developmental defects [22]. It is unknown what percentage of genes in a genome is actually sensitive to increased dosage.
In a diploid genome, decrease in the copy number of a gene can also have negative effects. Such genes are called “haploinsufficient” and require the presence of two wild type copies for full function. A systematic screen in yeast indicated that, under optimal culture conditions, ~3% of 6,000 yeast genes cause growth defect when present in single copy [23]. This is likely an underestimation, since many genes may be haploinsufficient in conditions that are not tested. Indeed, a study that assessed genome instability reported additional haploinsufficient genes [24]. Haploinsufficient genes are enriched in genome ontology categories relating to transcription and transcript processing, protein folding, protein transport, and ribosome biogenesis [23]. In humans, haploinsufficiency of tumor suppressors are linked to cancers (reviewed in [25]). Having a single copy of a tumor suppressor not only reduces the dosage of the tumor suppressor, but also predisposes the organism to recessive mutations [26]. For example, single copy of BRCA1 gene leads to reduced levels of BRCA1 protein which causes problems in DNA repair [27], and leads to increased susceptibility to mutations that can eliminate the remaining copy (reviewed in [28]).
Effect of multiple genes' dosage
Duplication or deletion of small chromosomal regions lead to copy number variations (CNVs), and change the dosage of single or multiple genes (Figure 1A). Some CNVs are associated with diseases, in particular neurological ones (reviewed in [29]). Duplication or deletion of larger chromosomal segments result in partial aneuploidies. Systematic analyses of partial aneuploidies in flies [30] and yeast [3] have shown a positive correlation between the size of the affected chromosomal segment and its influence. This result suggests that a change in copy number of multiple genes has a cumulative effect on the organism.
Genomic changes that lead to dosage differences. A) Duplication or deletion of different sizes of chromosomal segments are depicted. B) In a diploid species with X chromosomes, males contain a single X chromosome compared to two copies of each autosome. In species where Y chromosome does not contain many X-linked gene alleles (e.g. human), or in species where Y chromosome is nonexistent (e.g. C. elegans), X is monosomic in males.
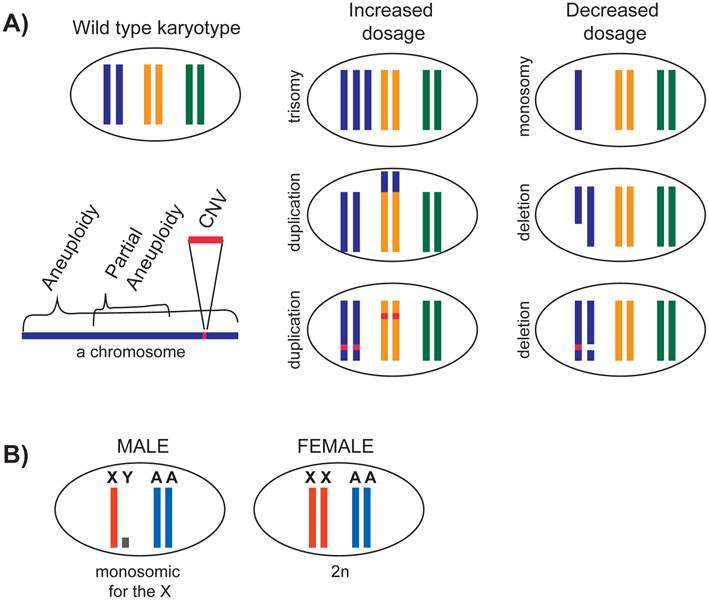
While CNVs and partial aneuploidies are fairly pervasive in a population, aneuploidies of whole chromosomes are often lethal in animals (reviewed in [2, 31]). In organisms that were studied, monosomy in a diploid organism (2n-1) is almost always lethal (reviewed in [32]). In D. melanogaster, monosomy of chromosome 4 is tolerated because this chromosome is small, heterochromatic, and contains few genes. In general, trisomy is better tolerated than monosomy. In humans, trisomy of 21 is viable and causes Down syndrome. Down syndrome phenotypes are thought to be caused by the cumulative effect of increased chromosome 21 gene dosage, however the entirety of the genes responsible for the phenotypes remain unclear (reviewed in [33], and [34]). Overall, the phenotype of partial and full aneuploidies and CNVs depend on the number and the types of genes that it contains.
In addition to causing problems based on the function of individual genes, in many species common stress pathways are activated as a general response to aneuploidy [3, 35]. In yeast, trisomy of several different chromosomes causes proteotoxic stress due to increased pressure on the protein degradation pathways to eliminate the extra proteins [11, 36]. The observation that aneuploidies trigger common response pathways may contribute to the difficulty of identifying those individual genes whose altered dosage can explain all Down syndrome phenotypes [37, 38].
While detrimental for development, aneuploidy is a hallmark of cancer cells (reviewed in [39]). There are several mechanisms by which cancer cells may tolerate aneuploidy (reviewed in [1]). Briefly, aneuploidy may not be detrimental to the proliferation of individual somatic cells compared to development of a whole organism. In addition, cancer cells often harbor decreased dosage of tumor suppressors genes (e.g. pRb and p53) and increased dosage of oncogenes (e.g. myc). Thus, gene dosage may act as a selection mechanism for those cancer cells with a growth advantage.
Monosomy of the X and the evolution of X chromosome dosage compensation
The X chromosome presents a natural case of aneuploidy in males. Males contain a single X chromosome and two copies of each autosome (XY, AA). In contrast, females contain a full set of chromosomes (XX, AA) (Figure 1B). In mammals, Y chromosome contains few genes, thus X chromosome is essentially monosomic in males. X is certainly monosomic in the case of C. elegans, where males do not contain any Y chromosome (XO). How is monosomy of the X tolerated in males?
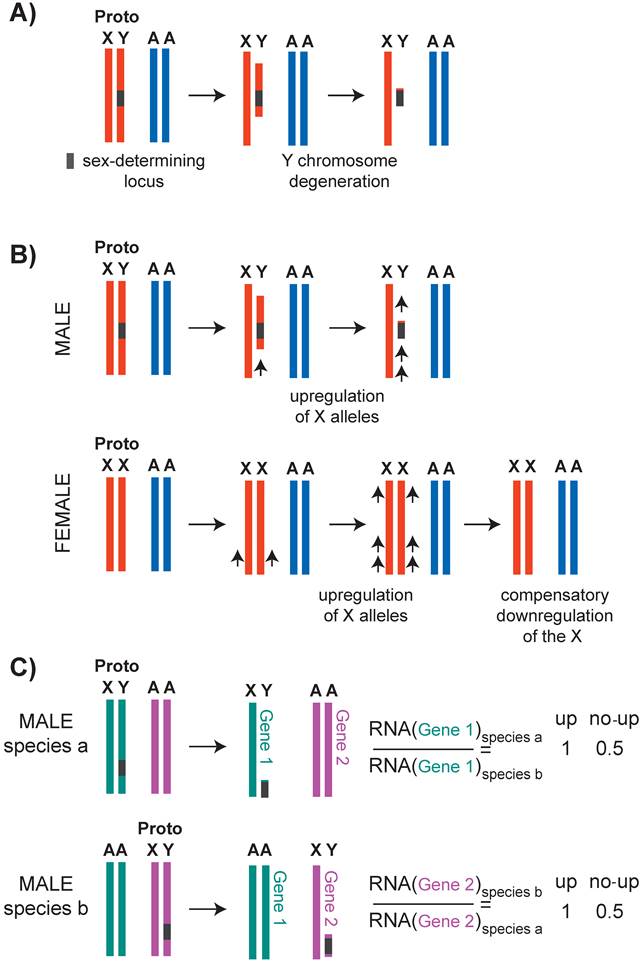
This question should be considered in the context of X chromosome evolution. X and Y chromosomes evolved from a pair of autosomes by a series of events that linked a sex determination locus to one sex by suppressing recombination between the two homologs (Figure 2A) (reviewed in [40, 41]). Lack of recombination is thought to result in Y chromosome degeneration, leaving most genes on the X chromosome in single copy in males. Susumu Ohno hypothesized that potential haploinsufficiencies unveiled by male monosomy were counteracted by increased expression from the single X chromosome (Figure 3).
Evolution of the X chromosomes, and Ohno's hypothesis. A) It is hypothesized that the sex chromosomes evolved by formation of a sex locus on the Y chromosome followed by suppressed recombination around this locus. With time, Y chromosome slowly degenerated. B) Upper panel: Ohno hypothesized that due to Y chromosome degeneration, the remaining alleles on the X chromosome became potentially haploinsufficient. To compensate for this, the alleles on the X chromosome were transcriptionally upregulated. Lower panel: It was also hypothesized that the upregulation of X alleles were not limited to males, and also occurred in females. This caused a potential overtranscription of the X-linked genes in females, therefore female-specific downregulation occurred. C) To test if X-upregulation occurred, one should compare ancestral (autosomal) and present level of X-linked gene expression. Since this is not possible, assuming that the function and expression of 1:1 orthologs are conserved, one can compare expression of 1:1 orthologs that are differentially located on X or autosomes. Recent studies on Ohno's hypothesis suggest that X-uprgeulation is only one of the several mechanisms of dealing with potential X haploinsufficieny, and not all genes were upregulated (see text).
X chromosome dosage compensation strategies in mammals, flies and worms. In flies, a male-specific dosage compensation complex increases X chromosome transcription in males, compensating both for potential monosomy of the X with respect to autosomes, and for X chromosome dosage difference between XY males and XX females. In mammals and in worms, X was hypothesized to be upregulated in males to counteract potential X monosomy in XO males. In females, upregulation of the X was counteracted by female-specific dosage compensation mechanisms. In mammals, X inactivation silences one of the X chromosomes in XX females to equalize X dosage between XY males and XX females. In worms both X chromosomes are downregulated by a factor of two in hermaphrodites to equalize X chromosome dosage between XX hermaphrodites and XO males. Note that X-upregulation did not apply to all genes (see Ohno's hypothesis section in the text).
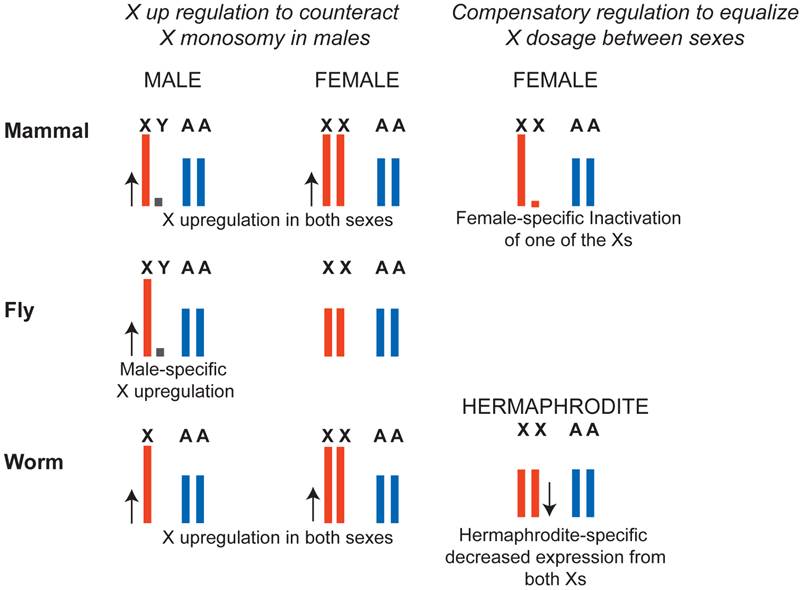
In D. melanogaster, transcription from the X chromosome in XY males is upregulated approximately two-fold, solving the potential haploinsufficiency of X-linked genes in males (Figure 3). In case of D. melanogaster, X-upregulation is male-specific and is accomplished by a well-defined dosage compensation complex. In mammals and C. elegans, it was hypothesized that upregulation of X-linked genes was not male-specific and, instead occurred in both sexes. This solved the problem of X monosomy in males, but increased X expression in females above autosomal levels. To compensate for this, female-specific dosage compensation mechanisms have evolved (Figure 3). In mammals, female-specific dosage compensation transcriptionally silences one of the X chromosomes (X inactivation). In worms, hermaphrodite-specific dosage compensation represses both X chromosomes in XX hermaphrodites by two-fold.
Ohno's hypothesis has received renewed attention in the past few years (reviewed in [42]). Evidence for X upregulation in mouse and C. elegans initially came from the observation that in males, the average level of transcripts from the single X chromosome and the two-copy autosomes are similar [43]. In addition, X transcript levels increase to above autosomal levels in the absence of female-specific dosage compensation in mouse [44] and C. elegans [45]. Also in support of X-upregulation in mice, the single X chromosome in males and the active X chromosome in females have higher RNA Pol II recruitment to promoters compared to autosomes [46, 47]. Comparison of overall transcription from the X and autosomes is problematic, because X chromosome and autosomal gene content differs (reviewed in [48, 49]). Therefore, to test Ohno's hypothesis, ideally one should compare expression levels of the ancestral and current X-linked genes. Since this is not possible, expression of an X-linked gene in one species may be compared to a one-to-one ortholog that resides on an autosome in another species (Figure 2C). Using this approach, a recent study found that the X chromosome is largely not upregulated in mice and humans [50]. A similar conclusion was reached for the C. elegans X chromosome [51].
How can we reconcile different lines of evidence for support and refusal of Ohno's hypothesis? It appears that X-upregulation did not happen across all the genes on the X chromosome. In mammals, dosage sensitive genes, such as those that belong to multi-subunit protein complexes were upregulated compared to dosage insensitive genes [52]. This suggests that a subset of X-linked genes is upregulated. In humans, an alternative way to deal with decreased dosage of an X-linked gene may have involved downregulation of autosomal genes that are within the same protein-protein interaction network [50]. In C. elegans, orhologs of yeast haploinsufficient genes are depleted from the X chromosome [51], suggesting that another way to solve potential haploinsufficiency is to move a dosage sensitive gene to an autosome. Therefore, Ohno's hypothesis of X-upregulation is one of many different mechanisms that counteracted the potential haploinsufficiency of individual X-linked genes in males.
Mechanisms of sex-specific X chromosome dosage compensation
The molecular mechanisms of X chromosome dosage compensation are studied mostly in the model organisms C. elegans, D. melanogaster, and Mus musculus. Although the problem of X chromosome dosage is the same in each of the three model organisms, molecular mechanisms of dosage compensation are different (Figure 3). In mammals, X inactivation machinery transcriptionally silences one of the X chromosomes in XX females. In flies, the male specific lethal (MSL) complex increases transcription from the single X chromosome by two-fold in XY males. In worms, the dosage compensation complex (DCC) halves transcription from both X chromosomes in XX hermaphrodites.
In recent years, research on the molecular mechanisms of the three dosage compensation systems revealed significant parallels in how dosage compensation machineries target the X chromosome, how they spread in cis, and how they regulate chromatin structure and transcription. Insights into these chromosome-wide processes were afforded by the genomic techniques that measure expression, binding and chromatin structure across the X chromosome. Below, I will discuss the three common themes in X chromosome dosage compensation: recruitment, spreading and regulation of chromatin structure.
Common themes: Recruitment of the dosage compensation machinery to the X chromosome
Initiation of X inactivation in mammals
X inactivation is mostly studied in placental mammals including mouse and human. Of the other two mammalian lineages, monotremes (e.g. platypus) do not have extensive X inactivation [50, 53, 54], and marsupials (e.g. kangaroo) show paternally imprinted X inactivation [50, 55-60]. In mice and humans, the mechanisms of X inactivation are generally conserved with a few notable differences. In mice, the paternal X chromosome is inactivated in the early embryo, and remains inactivated for the rest of embryogenesis in extraembryonic tissues such as the placenta [61-63]. In the inner cell mass of the mouse blastocyst, which includes cells that give rise to the embryo proper, the paternal X chromosome is reactivated and then either maternal or paternal X chromosome is randomly chosen for inactivation (random X inactivation) [64, 65]. In humans, paternal and maternal X chromosomes are both active until random X inactivation occurs in late blastocyst stage ([66], reviewed in [67]).
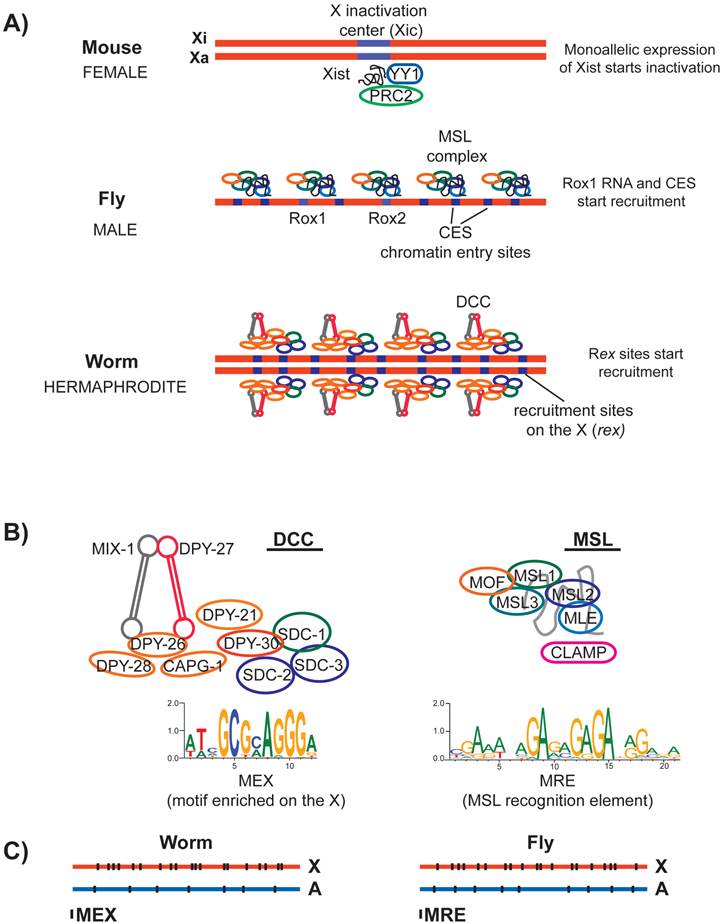
In mammals, random X inactivation targets one of the two X chromosomes. This is accomplished by the mono allelic transcription of a long non-coding RNA named Xist (X-inactive specific transcript) (reviewed in [68]). Xist is transcribed from a ~500 kb locus on the X chromosome, called the X inactivation center (Xic) (Figure 4A). Regulatory steps that lead to mono allelic transcription of Xist include other noncoding RNAs from Xic (reviewed in [69, 70]). Sex-specificity and randomness of Xist transcription are thought to be mediated by communication between the two Xic loci on the two homologs [71-73]. Deletion of the Xist locus eliminates X inactivation [74, 75], and insertion of the Xist locus to an autosomal site leads to ectopic silencing in cis [76-78]. Thus Xist is both necessary and sufficient for initiation of X inactivation.
Recruitment of the dosage compensation complexes in mammals, flies and worms. A) In mouse, one of the homologs is inactivated (Xi) and the other remains active (Xa). X inactivation is initiated randomly by monoallelic expression of a long noncoding RNA named Xist from the X chromosome that is destined to be inactivated. Xist may interact and recruit multiple proteins to the X. In flies, male specific dosage compensation complex (MSL) is assembled with either of the two noncoding RNAs named roX1 and roX2 at their loci. MSL complex is recruited to the X chromosome at a number of chromatin entry sites (CES). In worms, the hermaphrodite specific dosage compensation complex (DCC) is initially recruited to the X chromosome at a number of recruitment sites on the X (rex). B) Left panel: The core of the worm DCC is a 5-subunit condensin complex that shares 4/5 subunits with the canonical condensin I. At least five additional proteins interact with the condensin core and have a role in dosage compensation. A 12-bp motif (MEX) is enriched at the DCC recruitment sites on the X, and is shown below. Right panel: Subunits of the MSL complex and the structural noncoding RNA (roX) is shown in grey. A DNA sequence motif called MRE is enriched under the MSL recruitment sites on the X chromosome. CLAMP is required for MSL recruitment to the X. C) Both MEX and MRE are enriched on their respective X chromosomes, but there are many motifs present on the autosomes that are not bound by the dosage compensation complexes.
Recruitment of the MSL complex to the X chromosome in D. melanogaster
In D. melanogaster, the Male Specific Lethal (MSL) complex specifically binds to and upregulates transcription from the X chromosome in XY males (reviewed in [79, 80]). Sex-specificity of the MSL complex is provided by the MSL2 protein, which is expressed only in male embryos [81]. MLS2 binds to and stabilizes MSL1, and form a complex with MSL3, MLE RNA helicase (maleless), MOF histone acetyltransferase (males absent on the first), and one of two non-coding RNAs called roX1 and roX2 (RNA on the X) [82-85]. Ectopic insertion of roX RNA on autosomes recruits the MSL complex in cis to autosome, and in trans to the X chromosome [86-88].
Chromatin immunoprecipitation coupled with microarray analysis (ChIP-chip) and high-throughput sequencing (ChIP-seq) of MSL subunits found ~200 sites that direct initial recruitment of MSL to the X chromosome. These sites are called the chromatin entry sites (CES) or high affinity sites (HAS) (Figure 4A) (reviewed in [89]). The sequence of roX RNAs is not complementary to CES sequences on the X. Therefore, rather than specifying recruitment by a RNA-DNA hybridization event, the function of roX RNAs is likely structural [90-92]. In agreement with this, while the absence of both roX RNAs abolishes MSL localization to the X chromosome [93], overexpression of MSL1 and MSL2 can partially overcome the necessity for roX RNAs [94]. CESs contain a GA rich DNA sequence motif, named the MSL recognition element (MRE) (Figure 4B) [95-97]. Insertion of ~1 kb DNA fragments containing wild type MRE motif into an autosome leads to ectopic recruitment of the MSL complex [95]. A recently identified zinc finger protein, CLAMP binds to MRE, and is required for recruitment of MSL complex to the X chromosome [98].
Recently, comparative analysis of MSL binding in different Drosophila species provided additional insights into the mechanisms of recruitment. In several Drosophila species, multiple chromosomal translocations have formed neo-X and neo-autosomal regions [99, 100]. Analysis of MSL binding sites in Drosophila miranda suggests that while majority of newly formed CESs on the neo-X chromosome are due to mutations, about a third are formed by a mutant copy of a transposon carrying a MRE-like motif [101]. This study illustrates how transposons could provide a mechanism for coordinated recruitment of a chromatin-modifying complex to a large chromosomal domain.
Recruitment of the Dosage Compensation Complex to the X chromosome in C. elegans
In C. elegans, the Dosage Compensation Complex (DCC) binds to and represses both X chromosomes in XX hermaphrodites by an average of two-fold (Figure 4A) (reviewed in [102, 103]). At the core of the DCC is a specialized condensin complex. Condensins are evolutionarily conserved five-subunit protein complexes that are essential for proper chromosome condensation and segregation (reviewed in [104]). In metazoans, two types of condensin complexes (named I and II) share two Structural Maintenance of Chromosomes (SMC) protein subunits, and a set of three different non-SMC subunits [105]. The condensin core of the DCC shares four out of five subunits with condensin I, but includes an SMC variant called DPY-27 [106].
DPY-27 interacts with at least five other non-condensin proteins including SDC-1, SDC-2, SDC-3, DPY-30 and DPY-21 (Figure 4B) [107-112]. Sex-specificity of the DCC is provided by SDC-2 protein, which is expressed only in XX hermaphrodites during early embryogenesis [113]. SDC-2, SDC-3 and DPY-30 are required for the recruitment of the condensin portion of the DCC to the X chromosome [112].
C. elegans DCC first binds to a number of recruitment sites on the X (rex), and then spreads onto the X chromosome [102, 106]. There are approximately 100 predicted rex sites along the length of the 17.5 Mb X-chromosome. Initially, rex sites were identified by assaying the ability of DNA fragments, in the form of multi-copy extrachromosomal arrays to recruit the DCC [114]. ChIP-chip analysis of DCC identified additional recruitment sites, and defined a 10 bp DNA sequence motif that is enriched at the rex sites (Figure 4B) [115]. This motif was later extended to 12-bp and named the motif enriched on the X (MEX) [116, 117]. Although a 35 bp DNA fragment containing the motif as shown to recruit the DCC on extrachromosomal arrays [118], it is still unknown if the same fragment could recruit as a single copy insertion on an autosome. Nevertheless, the extrachromosomal recruitment assays show that MEX is important, because mutation of the motif resulted in loss of DCC recruitment [117, 118]. It is not known if any of the DCC subunits bind directly to MEX. Therefore, it remains unclear which proteins specify X-recruitment of the DCC via interaction with the MEX motif.
Parallels in specification of X-recruitment in C. elegans and D. melanogaster
DCC and MSL recruitment to the X chromosome show many parallels. The D. melanogaster MRE and C. elegans MEX motifs are not specific to the X chromosome and are only slightly enriched on their respective X chromosomes. D. melanogaster MRE is enriched on the X ~2-4-fold [95]. The C. elegans MEX is also ~2-4 fold enriched and slightly more clustered on the X chromosome (Figure 4C) [115, 117]. Presence of many unbound motifs on the autosomes in both species suggests that although critical, DNA sequence motifs cannot fully explain the X specificity of dosage compensation complex recruitment.
Specification of binding sites is a general biological problem common to all Transcription Factors (TFs). Many genome-wide binding studies show that TFs occupy only a small fraction of their potential targets. One contributor to specification of TF binding sites is the accessibility of the TF DNA sequence motifs to the respective TFs. TF binding motifs that are located at active promoters and enhancers that have lower nucleosome density are more likely to be bound [119-124]. In the case of D. melanogaster, it was shown that the MSL complex prefers more accessible binding sites for recruitment [125].
Although chromatin accessibility could play a role in the choice of binding motifs within the X chromosome, it cannot be a large factor in X-specification. Accessible recruitment motifs are present on both X and autosomes, but the dosage compensation complexes do not bind autosomes. Therefore, X-specific recruitment should involve other mechanisms that act to increase the usage of the motifs on the X. These mechanisms may involve cooperative recruitment by long-range interactions over long distance, or by increased concentration of the dosage compensation complexes on the X chromosome due to spreading, as discussed below.
Common themes: Dosage compensation complex spreading on the X chromosome
After X-specific recruitment, dosage compensation complexes spread in cis along the X chromosome. This recruitment-and-spreading type of mechanism may have evolved to regulate genes that translocate to the X chromosome. Spreading is also seen for many chromatin proteins involved in domain-wide gene regulation, such as HP1, Sir complex, and Polycomb complexes (reviewed in [126]).
Xist spreading on the X chromosome
After initiation of X chromosome inactivation, Xist and its associated proteins, including YY1 transcription factor [70] and the Polycomb Repressive Complex 2 (PRC2) [127] spread along the X (Figure 5A). Genomic techniques that mapped Xist localization at high resolution suggested that spreading occurs in two-steps: first to a smaller subset of Xist binding sites, and then to the remaining sites along the X (CHART-seq, [128], RAP-seq, [129]). The initial sites of spreading tend to be gene-rich regions and those that are physically close to the Xist locus. When Xist was inserted onto an autosome, autosomal spreading was not as high as on the X, suggesting that the X chromosome is more conducive to Xist spreading [130, 131]. High-resolution mapping of long-range chromosomal interactions showed that the inactivated X chromosome conforms into a Xist dependent three-dimensional structure [132]. This structure may be a factor in Xist spreading and/or maintenance of Xist binding (Figure 5B) [128, 129].
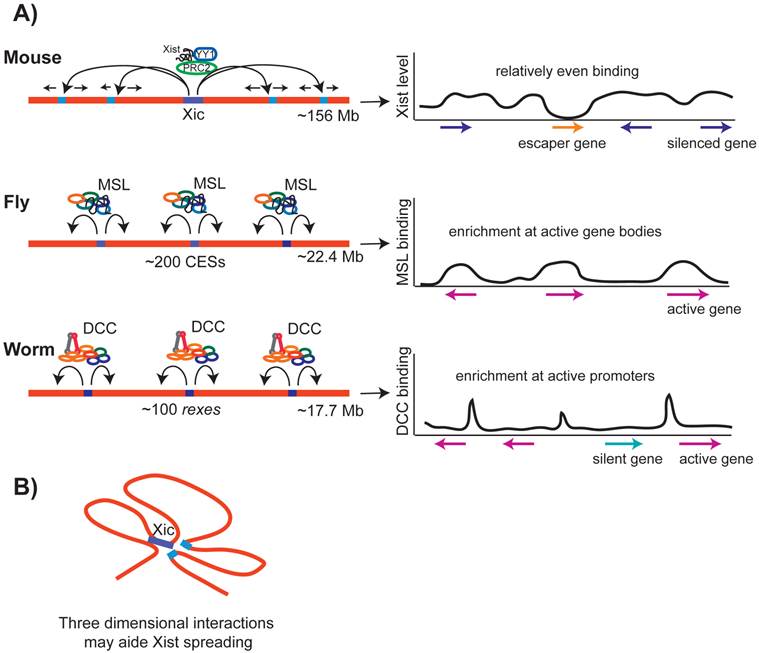
Spreading of the dosage compensation complexes in mammals, flies and worms. A) In mammals, Xist spreads initially to a number of loci, and then spreads along the rest of the X chromosome, binding at genes that are silenced. In flies, after recruitment to the CES sites, MSL complex spreads onto the X chromosome, preferentially accumulating towards the 3' end of transcribed regions of active genes. In worms, after recruitment to the rex sites, the DCC spreads onto the X chromosome, preferentially accumulating at a subset of active gene promoters and enhancers. B) In mammals, recent studies suggest that Xist spreading along the X chromosome may be aided by three-dimensional interactions between distant loci.
MSL spreading on the X chromosome in D. melanogaster
In flies, after initial recruitment to the CES sites, MSL spreads along the X (Figure 5A). Ectopic recruitment of MSL complex to an autosome lead to spreading [95], and an autosomal gene inserted onto the X chromosome recruited MSL [133], suggesting that MSL can spread onto autosomal genes. MSL spreads primarily to active gene bodies with a preferential binding at the 3' of transcribed regions (Figure 5C). MSL binding at active gene bodies is accomplished in part by the affinity of MSL3 to H3K36 trimethylated nucleosomes that are enriched at the 3' of transcribed regions [134, 135]. There may be two different modes of MSL binding, one at the recruitment sites and the other at the sites of spreading [136]. At the recruitment sites, MSL binds to DNA, and at gene bodies it binds to nucleosomes. The molecular mechanism by which MSL spreads from the recruitment sites to active gene bodies remains unclear.
DCC spreading on the X chromosome in C. elegans
In worms, after initial recruitment to rex sites, the DCC spreads along the X chromosome. Similar to MSL complex spreading in flies, DCC spreading is not X-sequence specific. DCC was shown to spread from the X chromosome into the autosomal region of an X;A fusion chromosome [116]. DCC spreads preferentially to transcriptionally active promoters and putative enhancers (Figure 5C) [115, 116, 137]. Low levels of DCC binding at active autosomal promoters was observed by ChIP-chip, suggesting that the DCC has some intrinsic affinity to active promoters regardless of the chromosome context [117]. This intrinsic affinity must be low, as ChIP-chip and ChIP-seq analyses with higher detergent concentration do not show significant autosomal binding [115, 116, 137]. Spreading may be a general feature of SMC containing protein complexes, as ectopic recruitment of yeast condensin lead to spreading to a nearby active promoter [138]. In yeast, it was suggested that the cohesin complex spreads by being pushed along the chromatin by RNA Polymerase II [139, 140]. The molecular mechanism by which the DCC and yeast condensin spread is unknown.
C. elegans condensin II was found to bind chromosomes in a manner similar to the DCC, showing ChIP-seq enrichment at a subset of active promoters and enhancers [137]. This may be a conserved property of condensins because similar binding patterns were observed for condensins in yeast [138], chicken cells [141], fly tissue culture cells [142] ,and in mouse embryonic stem cells [143]. In vitro, condensins bind to both naked DNA and chromatin non-specifically [146, 147]. In vivo, condensin binding sites may be specified by specific recruitment and certain features of chromatin structure. Indeed, yeast condensin was shown to preferentially bind H2A and H2A.Z [144]. A role for H2A.Z in DCC binding was also proposed based the observation that DCC immunofluorescence signal diffused off of the X chromosome upon H2A.Z knockdown [145]. Not all DCC sites contain H2A.Z, thus specificity of binding to chromatin remains unclear.
Common themes: Transcriptional regulation by altering chromatin structure of the X chromosome
Dosage compensation complex spreading leads to changes in X chromosome chromatin structure and transcription. In mammals, Xist spreading initiates X inactivation process, which involves compaction of the inactivated X chromosome into a cytogenetically observable Barr body. In flies and worms, X chromosome dosage compensation complexes regulate chromatin structure more slightly to “fine-tune” transcription.
Regulation of mammalian X chromosome structure
The process of X inactivation starts with Xist spreading, followed by gradual accumulation of heterochromatin-associated marks on the X chromosome (Figure 6). These include H3K27me3, H3K9me3, H4K20me3, macroH2A, and DNA methylation (reviewed in [80, 148]). This heterochromatin state maintains X inactivation, since Xist was shown to be important for initiation of silencing but not maintenance [74, 76, 149, 150]. It is not clear how these heterochromatic histone modifications are targeted to the X chromosome. For H3K27me3, it is thought that PRC2 is recruited to the X by binding to the Xist RNA [127, 151]. Paradoxically, immunofluorescence analysis of PRC2 and Xist do not show strong co-localization [152]. In addition, the presence of PRC2 and H3K27me3 does not explain all X inactivation [153, 154]. Given that additional histone marks, DNA modifications, and proteins such as HBiX1 and SMCHD1 are enriched on the X [155], it is possible that collective action of multiple repression mechanisms are needed to inactivate the X chromosome.
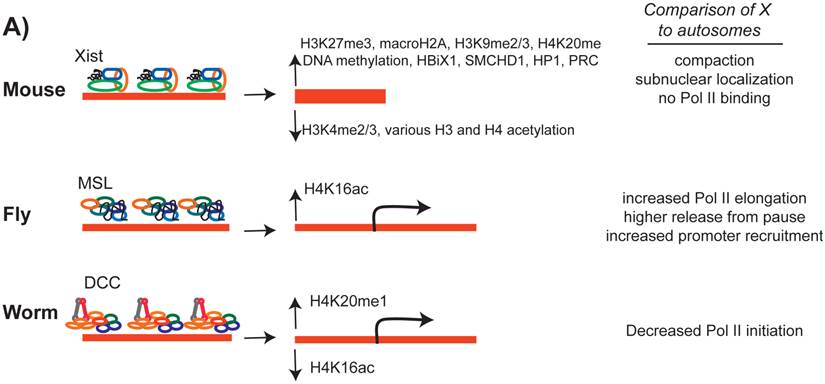
The effect of the dosage compensation complex on the X chromosome chromatin structure. In all three cases, a number of histone modifications are increased and some modifications are decreased on the X chromosome. While X inactivation silences most of X-linked genes, in C. elegans and D. melanogaster, transcription from the X chromosome is regulated by an average of two-fold.
X inactivation does not silence all genes. In mice, ~3% of alleles on the inactivated X chromosome show some level of transcription [156]. Escape from X inactivation is more pervasive in humans, and was estimated at ~15% [157]. The amount of escape per gene varies, as does the tissues in which escape occurs (reviewed in [158]). Proper expression of genes that escape X inactivation must be important in XX females because XO females with Turner syndrome show numerous deleterious phenotypes. In addition, increased expression of escaper genes might cause problems associated with Klinefelter syndrome (XXY) patients. It was proposed that genes escape X inactivation by being outside the Xist domain [159]. A recent study tested this hypothesis in mouse and showed that genes within the Xist domain can escape inactivation [160]. The presence of escaper genes within the Xist domain suggests that X inactivation might act on individual genes. In this case, enrichment of heterochromatin marks on the inactivated X chromosome may be a consequence of having many genes being silenced on the X.
Regulation of X chromosome structure by the MSL complex in D. melanogaster
MSL complex includes the MOF acetyltransferase, which catalyzes and increases the level of H4K16ac on the male X chromosome (Figure 6) [85, 161, 162]. MOF is present in both males and females, and has a general role in gene regulation genome-wide [163]. Within the MSL complex, MOF is targeted to transcriptionally active gene bodies and acetylates H4K16 [164-166]. Since H4K16ac is enriched in active gene bodies, it was hypothesized that H4K16ac may loosen nucleosome compaction [167, 168] and allow for more efficient transcriptional elongation [162].
Genome-wide analysis of engaged RNA Pol II levels by global run-on assay (GRO-seq) [169], ChIP-seq analysis of initiating and elongating forms of RNA Pol II [170], and analysis of 5' short RNA levels (5' CAP-seq, nascent-seq) [171] support the hypothesis that MSL increases transcriptional elongation. Also supporting this hypothesis is the observation that an elongation factor mutation reduces MSL-dependent upregulation of a dosage compensation reporter gene [172]. Current models of increased elongation by MSL include faster clearing of nucleosomes due to H4K16ac (reviewed in [80]) and/or reduction in negative DNA supercoiling [174-176]. One ChIP-seq study suggested that the MSL complex mediates ~1.2 fold increase in RNA Pol II recruitment to X-chromosome gene promoters in males [173]. A recent study that collectively analyzed various genome-wide data suggested that the MSL complex mediates more efficient release of paused RNA Pol II in addition to increasing transcriptional elongation on the male X chromosome [171].
MSL complex may also affect higher order chromosome structure, since MSL recruitment sites are closer to each other within the nucleus in males compared to females [177]. Although a previous study suggested that nuclear pore proteins may be involved in regulation of higher order structure of the male X chromosome [178], another study reported that nuclear pore components do not affect X chromosome structure as measured by the distance between MSL recruitment sites [177]. The function of MSL-mediated higher-order chromosome structure in fly dosage compensation remains unclear.
Regulation of X chromosome structure and transcription by C. elegans DCC
DCC regulates X chromosome structure, in particular by increasing H4K20me1 levels along the X [179, 180]. GRO-seq analysis upon DCC depletion indicate that the DCC reduces RNA Pol II levels at the X chromosome promoters [45]. It is not clear how H4K20me1 enrichment may reduce RNA Pol II recruitment to X-linked promoters. DCC mediated H4K20me1 enrichment on the X leads to H4K16ac depletion [181]. It was hypothesized that H4K20me1 and H4K16ac act antagonistically [182, 183], perhaps to regulate dynamics of RNA Pol II pausing [184]. However, this model of regulation does not fit the DCC, because there is no widespread RNA Pol II pausing in C. elegans, and transcriptional repression occurs at the level of Pol II recruitment to promoters [45].
H4K20me1 is highly dynamic with respect to cell cycle (Reviewed in [185]). Immunofluorescence and western blot analyses in mammalian tissue culture cells showed that H4K20me1 levels increase during G2 through M phase, and then reduces dramatically after mitosis [186-190]. In contrast, H4K16ac is low during mitosis presumably because acetylation decreases H4-H2 interaction that mediates chromosome compaction [191]. Notably, increased H4K20me1 on mitotic chromosomes coincides with condensin binding. It is possible that the DCC mediates a “mitosis-like” chromosome structure on the C. elegans X chromosomes. While X-linked promoters were shown to support higher nucleosome occupancy, this is not specific to hermaphrodites [192]. Unfortunately, compaction of the X chromosomes has not been measured and compared to autosomes and mitotic chromosomes. It is also unclear how a mitosis-like chromosome structure causes a reduction in promoter recruitment of RNA Pol II.
Fine-tuning transcription by the dosage compensation complexes in D. melanogaster and C. elegans
Unlike X inactivation that silences majority of genes, fly and worm dosage compensation “fine-tune” transcription. The MSL complex activates and the DCC represses transcription from the X chromosomes by an average of two-fold. Comparing those X-linked genes whose expression decreases upon MSL knockdown to those genes that are bound by the MSL complex indicated a slight correlation between MSL binding and MSL-mediated change in transcription [166]. Approximately half of genes that were differentially expressed upon MSL knockdown were actually bound by the MSL complex [163]. The overall transcriptional effect of MLS2 and MOF knockdown in S2 cells was found to be on average ~1.35 fold [6, 193]. However, transcriptional changes upon knockdown experiments should be examined carefully; since ~16% of all genes are on the X chromosome, and misregulating X expression could have considerable indirect effects.
In C. elegans, expression analysis in hypomorphic mutants and upon RNAi depletion of sdc-2 and dpy-27 indicated a lack of correlation between DCC binding and DCC-mediated transcriptional repression [117]. The DCC binds to ~75% of expressed genes on the X chromosome [115, 116, 137]. Only half of the DCC bound genes are misregulated upon DCC knockdown, and half of misregulated genes are bound by the DCC. Although there was an average of two-fold upregulation across the X chromosome upon DCC depletion, the range of regulation was 1.5 up to 10 fold [117]. Since the X chromosome contains ~15% of all coding genes, secondary effects are likely to be confounding.
Unlike the MSL complex in flies, the DCC knockdown in worms caused widespread effects on autosomal transcription [117]. With respect to absolute number, microarray experiments revealed that both SDC-2 and DPY-27 knockdown affected more autosomal genes compared to X-linked genes. In contrast, the effect of MSL1 knockdown was mostly restricted to the X chromosome [163]. In addition, while knockdown of MSL complex caused an approximately equal number of autosomal genes to increase or decrease in transcription [163], the effect of DCC was asymmetrical. Approximately 4 times more autosomal genes decreased in expression as opposed to increased [117]. The molecular mechanisms of such genome-wide effect by the DCC remain unclear (reviewed in [194]).
Dosage Compensation and Development
Mechanisms that link dosage compensation to sex determination
In flies and worms, dosage compensation mechanisms are established during embryogenesis and are tightly linked to sex determination (Figure 7A). In mice and humans, sex determination is governed by the SRY (sex-determining region Y) gene on the Y chromosome (reviewed in [195]). Expression of SRY leads to male development, while the absence of SRY leads to female development ([196, 197]). X inactivation may be uncoupled from sex determination in mammals, as expression of SRY in XX mice caused male development in the presence of X inactivation [198].
The link between dosage compensation and development. A) Dosage compensation is integrated into the transcriptional networks that are required for proper development. In worms and flies, sex determination pathways control sex-specific recruitment of the dosage compensation complexes to the X chromosome. B) Recent studies suggest that transcriptional pathways that promote multipotency repress X inactivation, which promotes differentiation.
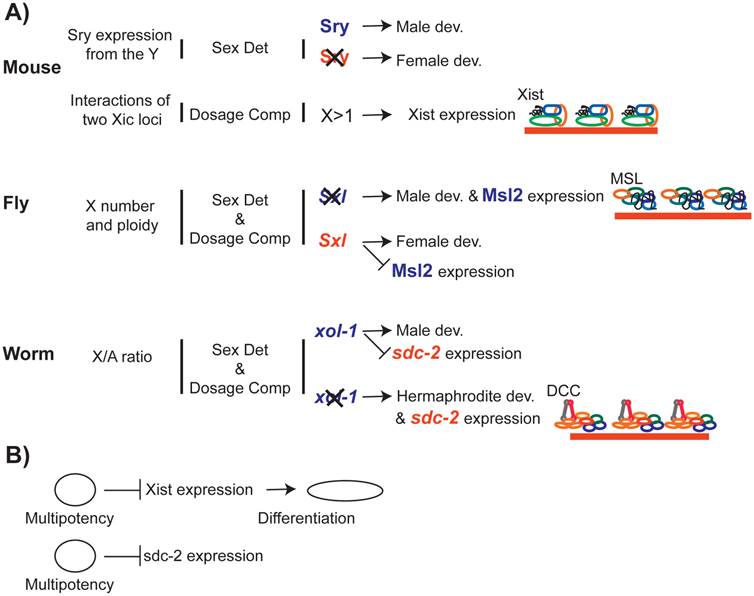
In contrast, dosage compensation depends on sex determination in C. elegans and D. melanogaster. In D. melanogaster, sex is determined by the sex lethal gene (Sxl) (reviewed in [199]). Sxl is transcribed in males, but contains a terminator in one exon. In females, this exon is spliced out, allowing for translation of a functional protein, SXL, which acts to repress MSL2 [200-203]. In males, the absence of SXL allows MSL2 expression, and thereby triggers dosage compensation [81]. Therefore, in D. melanogaster, sex determination pathway limits X chromosome dosage compensation to males.
In C. elegans, sex is determined by xol-1, a gene that promotes male development [204, 205]. A number of X-linked and autosomal genes [206-208] battle over xol-1 expression ([209, 210]). In males, XOL-1 represses sdc-2, which is required for both hermaphrodite development and recruitment of the DCC to the X chromosomes [205]. In the absence of SDC-2 in males, DCC does not bind and repress the X chromosome [113]. Therefore, in C. elegans the sex determination pathway limits dosage compensation to hermaphrodites.
Role of Dosage Compensation in Development
In C. elegans, D. melanogaster, mice, and humans, failure to dosage compensate is lethal during development. In mice, lack of X inactivation results in continual deterioration of the embryo and death around 10 days post coitum [211]. Dosage compensation mechanisms are also essential in D. melanogaster and C. elegans development ([211, 212]). In C. elegans, dosage compensation mutant hermaphrodites die late in embryogenesis or as early larvae [213-215]. It is still unknown if there are specific developmental processes that are significantly affected by a lack of dosage compensation.
Recent studies in mammals and C. elegans suggest that dosage compensation mechanisms are linked to differentiation (Figure 7B). In C. elegans, prolonged developmental plasticity due to mes-2 (PRC2) mutation caused a delay in DCC localization to the X chromosome because of a delay in SDC-2 expression [216]. In mammals, several genes important for maintenance of stem cell state repress Xist expression (reviewed in [217, 218]). In return, X inactivation causes repression of genes required for totipotency [219]. How X chromosome dosage compensation affects genome-wide transcriptional networks that regulate development and differentiation in the two sexes, remains an open question.
Conclusion and Perspective
Maintaining proper chromosome dosage is important for an organism's fitness. In many animals, evolution of the sex chromosomes resulted in males and females to have different X chromosome dosage. In response to this, X chromosome dosage compensation mechanisms have evolved. Amongst the three well-studied model systems (flies, worms and mammals), dosage compensation strategies are largely different. However, the molecular mechanisms of dosage compensation complexes do share many common characteristics that include specific recruitment to the X chromosome, cis-spreading along the X, and regulation of chromatin structure and transcription. It appears that in different organisms, different dosage compensation strategies coopted existing mechanisms of gene regulation to the X chromosome. Therefore, the mechanistic insights from dosage compensation studies will continue to contribute to our understanding of general mechanisms of transcription regulation.
In addition to mechanistic insights into gene regulation, dosage compensation studies are important because, X chromosome dosage compensation is an integral part of the transcriptional regulatory networks that ensure proper development and differentiation in many species. Yet, numerous questions remain about the evolution and function of X chromosome dosage compensation. For example, does dosage compensation have a role in sexual dimorphism? How does dosage compensation contribute to diversity of gene expression between individuals? What role does dosage compensation mechanisms play in evolution? Future research on X chromosome dosage compensation mechanisms in model and non-model organisms will help answer these important questions.
Acknowledgements
I thank Ercan lab members for valuable discussions and comments, and Sarah Albritton for her input on the manuscript. MRE motif logo was kindly provided by Marcela Soruco and Erica Larschan.
Competing Interests
The author has declared that no competing interest exists.
References
1. Torres EM, Williams BR, Amon A. Aneuploidy: cells losing their balance. Genetics. 2008;179:737-46 doi:10.1534/genetics.108.090878
2. Tang YC, Amon A. Gene copy-number alterations: a cost-benefit analysis. Cell. 2013;152:394-405 doi:10.1016/j.cell.2012.11.043
3. Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ. et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916-24 doi:10.1126/science.1142210
4. Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L. et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321-5 doi:10.1038/nature09529
5. Huettel B, Kreil DP, Matzke M, Matzke AJ. Effects of aneuploidy on genome structure, expression, and interphase organization in Arabidopsis thaliana. PLoS genetics. 2008;4:e1000226. doi:10.1371/journal.pgen.1000226
6. Gupta V, Parisi M, Sturgill D, Nuttall R, Doctolero M, Dudko OK. et al. Global analysis of X-chromosome dosage compensation. Journal of biology. 2006;5:3. doi:10.1186/jbiol30
7. Vacik T, Ort M, Gregorova S, Strnad P, Blatny R, Conte N. et al. Segmental trisomy of chromosome 17: a mouse model of human aneuploidy syndromes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4500-5 doi:10.1073/pnas.0500802102
8. Kahlem P, Sultan M, Herwig R, Steinfath M, Balzereit D, Eppens B. et al. Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of down syndrome. Genome research. 2004;14:1258-67 doi:10.1101/gr.1951304
9. Lyle R, Gehrig C, Neergaard-Henrichsen C, Deutsch S, Antonarakis SE. Gene expression from the aneuploid chromosome in a trisomy mouse model of down syndrome. Genome research. 2004;14:1268-74 doi:10.1101/gr.2090904
10. Altug-Teber O, Bonin M, Walter M, Mau-Holzmann UA, Dufke A, Stappert H. et al. Specific transcriptional changes in human fetuses with autosomal trisomies. Cytogenetic and genome research. 2007;119:171-84 doi:10.1159/000112058
11. Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Molecular systems biology. 2012;8:608. doi:10.1038/msb.2012.40
12. Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schutz F. et al. Segmental copy number variation shapes tissue transcriptomes. Nature genetics. 2009;41:424-9 doi:10.1038/ng.345
13. Slager J, Kjos M, Attaiech L, Veening JW. Antibiotic-induced replication stress triggers bacterial competence by increasing gene dosage near the origin. Cell. 2014;157:395-406 doi:10.1016/j.cell.2014.01.068
14. Hong J, Gresham D. Molecular specificity, convergence and constraint shape adaptive evolution in nutrient-poor environments. PLoS genetics. 2014;10:e1004041. doi:10.1371/journal.pgen.1004041
15. Claycomb JM, Orr-Weaver TL. Developmental gene amplification: insights into DNA replication and gene expression. Trends in genetics: TIG. 2005;21:149-62 doi:10.1016/j.tig.2005.01.009
16. Katz W, Weinstein B, Solomon F. Regulation of tubulin levels and microtubule assembly in Saccharomyces cerevisiae: consequences of altered tubulin gene copy number. Molecular and cellular biology. 1990;10:5286-94
17. Driever W, Nusslein-Volhard C. The bicoid protein determines position in the Drosophila embryo in a concentration-dependent manner. Cell. 1988;54:95-104
18. Hurles ME, Dermitzakis ET, Tyler-Smith C. The functional impact of structural variation in humans. Trends in genetics: TIG. 2008;24:238-45 doi:10.1016/j.tig.2008.03.001
19. Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223-41 doi:10.1016/j.cell.2012.02.039
20. Vacic V, McCarthy S, Malhotra D, Murray F, Chou HH, Peoples A. et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499-503 doi:10.1038/nature09884
21. Hollox EJ, Huffmeier U, Zeeuwen PL, Palla R, Lascorz J, Rodijk-Olthuis D. et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nature genetics. 2008;40:23-5 doi:10.1038/ng.2007.48
22. Hemmat M, Rumple MJ, Mahon LW, Strom CM, Anguiano A, Talai M. et al. Short stature, digit anomalies and dysmorphic facial features are associated with the duplication of miR-17 ~ 92 cluster. Molecular cytogenetics. 2014;7:27. doi:10.1186/1755-8166-7-27
23. Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW. et al. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169:1915-25 doi:10.1534/genetics.104.036871
24. Choy JS, O'Toole E, Schuster BM, Crisp MJ, Karpova TS, McNally JG. et al. Genome-wide haploinsufficiency screen reveals a novel role for gamma-TuSC in spindle organization and genome stability. Molecular biology of the cell. 2013;24:2753-63 doi:10.1091/mbc.E12-12-0902
25. Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nature reviews Genetics. 2012;13:189-203 doi:10.1038/nrg3123
26. Boone PM, Campbell IM, Baggett BC, Soens ZT, Rao MM, Hixson PM. et al. Deletions of recessive disease genes: CNV contribution to carrier states and disease-causing alleles. Genome research. 2013;23:1383-94 doi:10.1101/gr.156075.113
27. Konishi H, Mohseni M, Tamaki A, Garay JP, Croessmann S, Karnan S. et al. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17773-8 doi:10.1073/pnas.1110969108
28. Welcsh PL, King MC. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Human molecular genetics. 2001;10:705-13
29. Girirajan S, Campbell CD, Eichler EE. Human copy number variation and complex genetic disease. Annual review of genetics. 2011;45:203-26 doi:10.1146/annurev-genet-102209-163544
30. Lindsley DL, Sandler L, Baker BS, Carpenter AT, Denell RE, Hall JC. et al. Segmental aneuploidy and the genetic gross structure of the Drosophila genome. Genetics. 1972;71:157-84
31. Birchler JA. Aneuploidy in plants and flies: the origin of studies of genomic imbalance. Seminars in cell & developmental biology. 2013;24:315-9 doi:10.1016/j.semcdb.2013.02.004
32. Siegel JJ, Amon A. New insights into the troubles of aneuploidy. Annual review of cell and developmental biology. 2012;28:189-214 doi:10.1146/annurev-cellbio-101011-155807
33. Lana-Elola E, Watson-Scales SD, Fisher EM, Tybulewicz VL. Down syndrome: searching for the genetic culprits. Disease models & mechanisms. 2011;4:586-95 doi:10.1242/dmm.008078
34. Patterson D. Molecular genetic analysis of Down syndrome. Human genetics. 2009;126:195-214 doi:10.1007/s00439-009-0696-8
35. Sheltzer JM, Torres EM, Dunham MJ, Amon A. Transcriptional consequences of aneuploidy. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:12644-9 doi:10.1073/pnas.1209227109
36. Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP. et al. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71-83 doi:10.1016/j.cell.2010.08.038
37. Lyle R, Bena F, Gagos S, Gehrig C, Lopez G, Schinzel A. et al. Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. European journal of human genetics: EJHG. 2009;17:454-66 doi:10.1038/ejhg.2008.214
38. Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L. et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12031-6 doi:10.1073/pnas.0813248106
39. Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO reports. 2012;13:501-14 doi:10.1038/embor.2012.55
40. Charlesworth D, Charlesworth B, Marais G. Steps in the evolution of heteromorphic sex chromosomes. Heredity. 2005;95:118-28 doi:10.1038/sj.hdy.6800697
41. Vicoso B, Bachtrog D. Progress and prospects toward our understanding of the evolution of dosage compensation. Chromosome research: an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology. 2009;17:585-602 doi:10.1007/s10577-009-9053-y
42. Disteche CM. Dosage compensation of the sex chromosomes. Annual review of genetics. 2012;46:537-60 doi:10.1146/annurev-genet-110711-155454
43. Deng X, Hiatt JB, Nguyen DK, Ercan S, Sturgill D, Hillier LW. et al. Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nature genetics. 2011;43:1179-85 doi:10.1038/ng.948
44. Lin H, Halsall JA, Antczak P, O'Neill LP, Falciani F, Turner BM. Relative overexpression of X-linked genes in mouse embryonic stem cells is consistent with Ohno's hypothesis. Nature genetics. 2011;43:1169-70 author reply 71-2. doi:10.1038/ng.992
45. Kruesi WS, Core LJ, Waters CT, Lis JT, Meyer BJ. Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. eLife. 2013;2:e00808. doi:10.7554/eLife.00808
46. Deng X, Berletch JB, Ma W, Nguyen DK, Hiatt JB, Noble WS. et al. Mammalian X upregulation is associated with enhanced transcription initiation, RNA half-life, and MOF-mediated H4K16 acetylation. Developmental cell. 2013;25:55-68 doi:10.1016/j.devcel.2013.01.028
47. Yildirim E, Sadreyev RI, Pinter SF, Lee JT. X-chromosome hyperactivation in mammals via nonlinear relationships between chromatin states and transcription. Nature structural & molecular biology. 2012;19:56-61 doi:10.1038/nsmb.2195
48. Ellegren H, Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nature reviews Genetics. 2007;8:689-98 doi:10.1038/nrg2167
49. Parsch J, Ellegren H. The evolutionary causes and consequences of sex-biased gene expression. Nature reviews Genetics. 2013;14:83-7 doi:10.1038/nrg3376
50. Julien P, Brawand D, Soumillon M, Necsulea A, Liechti A, Schutz F. et al. Mechanisms and evolutionary patterns of mammalian and avian dosage compensation. PLoS biology. 2012;10:e1001328. doi:10.1371/journal.pbio.1001328
51. Albritton SE, Kranz AL, Rao P, Kramer M, Dieterich C, Ercan S. Sex-Biased Gene Expression and Evolution of the X Chromosome in Nematodes. Genetics. 2014;197:865-83 doi:10.1534/genetics.114.163311
52. Pessia E, Makino T, Bailly-Bechet M, McLysaght A, Marais GA. Mammalian X chromosome inactivation evolved as a dosage-compensation mechanism for dosage-sensitive genes on the X chromosome. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5346-51 doi:10.1073/pnas.1116763109
53. Deakin JE, Hore TA, Koina E, Marshall Graves JA. The status of dosage compensation in the multiple X chromosomes of the platypus. PLoS genetics. 2008;4:e1000140. doi:10.1371/journal.pgen.1000140
54. Livernois AM, Waters SA, Deakin JE, Marshall Graves JA, Waters PD. Independent evolution of transcriptional inactivation on sex chromosomes in birds and mammals. PLoS genetics. 2013;9:e1003635. doi:10.1371/journal.pgen.1003635
55. Mahadevaiah SK, Royo H, VandeBerg JL, McCarrey JR, Mackay S, Turner JM. Key features of the X inactivation process are conserved between marsupials and eutherians. Current biology: CB. 2009;19:1478-84 doi:10.1016/j.cub.2009.07.041
56. Gribnau J, Grootegoed JA. Origin and evolution of X chromosome inactivation. Current opinion in cell biology. 2012;24:397-404 doi:10.1016/j.ceb.2012.02.004
57. Cooper DW. Directed genetic change model for X chromosome inactivation in eutherian mammals. Nature. 1971;230:292-4
58. Sharman GB. Late DNA replication in the paternally derived X chromosome of female kangaroos. Nature. 1971;230:231-2
59. Livernois AM, Graves JA, Waters PD. The origin and evolution of vertebrate sex chromosomes and dosage compensation. Heredity. 2012;108:50-8 doi:10.1038/hdy.2011.106
60. Grant J, Mahadevaiah SK, Khil P, Sangrithi MN, Royo H, Duckworth J. et al. Rsx is a metatherian RNA with Xist-like properties in X-chromosome inactivation. Nature. 2012;487:254-8 doi:10.1038/nature11171
61. Huynh KD, Lee JT. Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature. 2003;426:857-62 doi:10.1038/nature02222
62. Patrat C, Okamoto I, Diabangouaya P, Vialon V, Le Baccon P, Chow J. et al. Dynamic changes in paternal X-chromosome activity during imprinted X-chromosome inactivation in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5198-203 doi:10.1073/pnas.0810683106
63. Takagi N, Sasaki M. Preferential inactivation of the paternally derived X chromosome in the extraembryonic membranes of the mouse. Nature. 1975;256:640-2
64. Mak W, Nesterova TB, de Napoles M, Appanah R, Yamanaka S, Otte AP. et al. Reactivation of the paternal X chromosome in early mouse embryos. Science. 2004;303:666-9 doi:10.1126/science.1092674
65. Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science. 2004;303:644-9 doi:10.1126/science.1092727
66. Moreira de Mello JC, de Araujo ES, Stabellini R, Fraga AM, de Souza JE, Sumita DR. et al. Random X inactivation and extensive mosaicism in human placenta revealed by analysis of allele-specific gene expression along the X chromosome. PloS one. 2010;5:e10947. doi:10.1371/journal.pone.0010947
67. Escamilla-Del-Arenal M, da Rocha ST, Heard E. Evolutionary diversity and developmental regulation of X-chromosome inactivation. Human genetics. 2011;130:307-27 doi:10.1007/s00439-011-1029-2
68. Augui S, Nora EP, Heard E. Regulation of X-chromosome inactivation by the X-inactivation centre. Nature reviews Genetics. 2011;12:429-42 doi:10.1038/nrg2987
69. Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308-23 doi:10.1016/j.cell.2013.02.016
70. Jeon Y, Lee JT. YY1 tethers Xist RNA to the inactive X nucleation center. Cell. 2011;146:119-33 doi:10.1016/j.cell.2011.06.026
71. Xu N, Tsai CL, Lee JT. Transient homologous chromosome pairing marks the onset of X inactivation. Science. 2006;311:1149-52 doi:10.1126/science.1122984
72. Masui O, Bonnet I, Le Baccon P, Brito I, Pollex T, Murphy N. et al. Live-cell chromosome dynamics and outcome of X chromosome pairing events during ES cell differentiation. Cell. 2011;145:447-58 doi:10.1016/j.cell.2011.03.032
73. Barakat TS, Loos F, van Staveren S, Myronova E, Ghazvini M, Grootegoed JA. et al. The trans-activator RNF12 and cis-acting elements effectuate X chromosome inactivation independent of X-pairing. Molecular cell. 2014;53:965-78 doi:10.1016/j.molcel.2014.02.006
74. Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996;379:131-7 doi:10.1038/379131a0
75. Marahrens Y, Panning B, Dausman J, Strauss W, Jaenisch R. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes & development. 1997;11:156-66
76. Wutz A, Jaenisch R. A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Molecular cell. 2000;5:695-705
77. Hall LL, Byron M, Sakai K, Carrel L, Willard HF, Lawrence JB. An ectopic human XIST gene can induce chromosome inactivation in postdifferentiation human HT-1080 cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8677-82 doi:10.1073/pnas.132468999
78. Jiang J, Jing Y, Cost GJ, Chiang JC, Kolpa HJ, Cotton AM. et al. Translating dosage compensation to trisomy 21. Nature. 2013;500:296-300 doi:10.1038/nature12394
79. Straub T, Becker PB. Dosage compensation: the beginning and end of generalization. Nature reviews Genetics. 2007;8:47-57 doi:10.1038/nrg2013
80. Ferrari F, Alekseyenko AA, Park PJ, Kuroda MI. Transcriptional control of a whole chromosome: emerging models for dosage compensation. Nature structural & molecular biology. 2014;21:118-25 doi:10.1038/nsmb.2763
81. Kelley RL, Solovyeva I, Lyman LM, Richman R, Solovyev V, Kuroda MI. Expression of msl-2 causes assembly of dosage compensation regulators on the X chromosomes and female lethality in Drosophila. Cell. 1995;81:867-77
82. Lyman LM, Copps K, Rastelli L, Kelley RL, Kuroda MI. Drosophila male-specific lethal-2 protein: structure/function analysis and dependence on MSL-1 for chromosome association. Genetics. 1997;147:1743-53
83. Gu W, Szauter P, Lucchesi JC. Targeting of MOF, a putative histone acetyl transferase, to the X chromosome of Drosophila melanogaster. Developmental genetics. 1998;22:56-64 doi:10.1002/(SICI)1520-6408(1998)22:1<56::AID-DVG6>3.0.CO;2-6
84. Copps K, Richman R, Lyman LM, Chang KA, Rampersad-Ammons J, Kuroda MI. Complex formation by the Drosophila MSL proteins: role of the MSL2 RING finger in protein complex assembly. The EMBO journal. 1998;17:5409-17 doi:10.1093/emboj/17.18.5409
85. Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, Allis CD. et al. The drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Molecular and cellular biology. 2000;20:312-8
86. Kelley RL, Meller VH, Gordadze PR, Roman G, Davis RL, Kuroda MI. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell. 1999;98:513-22
87. Park Y, Kelley RL, Oh H, Kuroda MI, Meller VH. Extent of chromatin spreading determined by roX RNA recruitment of MSL proteins. Science. 2002;298:1620-3 doi:10.1126/science.1076686
88. Park SW, Oh H, Lin YR, Park Y. MSL cis-spreading from roX gene up-regulates the neighboring genes. Biochemical and biophysical research communications. 2010;399:227-31 doi:10.1016/j.bbrc.2010.07.059
89. Chery J, Larschan E. X-marks the spot: X-chromosome identification during dosage compensation. Biochimica et biophysica acta. 2014;1839:234-40 doi:10.1016/j.bbagrm.2013.12.007
90. Maenner S, Muller M, Frohlich J, Langer D, Becker PB. ATP-dependent roX RNA remodeling by the helicase maleless enables specific association of MSL proteins. Molecular cell. 2013;51:174-84 doi:10.1016/j.molcel.2013.06.011
91. Ilik IA, Quinn JJ, Georgiev P, Tavares-Cadete F, Maticzka D, Toscano S. et al. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Molecular cell. 2013;51:156-73 doi:10.1016/j.molcel.2013.07.001
92. Park Y, Mengus G, Bai X, Kageyama Y, Meller VH, Becker PB. et al. Sequence-specific targeting of Drosophila roX genes by the MSL dosage compensation complex. Molecular cell. 2003;11:977-86
93. Meller VH, Rattner BP. The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. The EMBO journal. 2002;21:1084-91 doi:10.1093/emboj/21.5.1084
94. Oh H, Park Y, Kuroda MI. Local spreading of MSL complexes from roX genes on the Drosophila X chromosome. Genes & development. 2003;17:1334-9 doi:10.1101/gad.1082003
95. Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P. et al. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell. 2008;134:599-609 doi:10.1016/j.cell.2008.06.033
96. Dahlsveen IK, Gilfillan GD, Shelest VI, Lamm R, Becker PB. Targeting determinants of dosage compensation in Drosophila. PLoS genetics. 2006;2:e5. doi:10.1371/journal.pgen.0020005
97. Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB. The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS genetics. 2008;4:e1000302. doi:10.1371/journal.pgen.1000302
98. Soruco MM, Chery J, Bishop EP, Siggers T, Tolstorukov MY, Leydon AR. et al. The CLAMP protein links the MSL complex to the X chromosome during Drosophila dosage compensation. Genes & development. 2013;27:1551-6 doi:10.1101/gad.214585.113
99. Bachtrog D, Toda NR, Lockton S. Dosage compensation and demasculinization of X chromosomes in Drosophila. Current biology: CB. 2010;20:1476-81 doi:10.1016/j.cub.2010.06.076
100. Vicoso B, Bachtrog D. Reversal of an ancient sex chromosome to an autosome in Drosophila. Nature. 2013;499:332-5 doi:10.1038/nature12235
101. Ellison CE, Bachtrog D. Dosage compensation via transposable element mediated rewiring of a regulatory network. Science. 2013;342:846-50 doi:10.1126/science.1239552
102. Ercan S, Lieb JD. C. elegans dosage compensation: a window into mechanisms of domain-scale gene regulation. Chromosome research: an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology. 2009;17:215-27 doi:10.1007/s10577-008-9011-0
103. Wood AJ, Severson AF, Meyer BJ. Condensin and cohesin complexity: the expanding repertoire of functions. Nature reviews Genetics. 2010;11:391-404 doi:10.1038/nrg2794
104. Hirano T. Condensins: universal organizers of chromosomes with diverse functions. Genes & development. 2012;26:1659-78 doi:10.1101/gad.194746.112
105. Ono T, Losada A, Hirano M, Myers MP, Neuwald AF, Hirano T. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell. 2003;115:109-21
106. Csankovszki G, Collette K, Spahl K, Carey J, Snyder M, Petty E. et al. Three distinct condensin complexes control C. elegans chromosome dynamics. Current biology: CB. 2009;19:9-19 doi:10.1016/j.cub.2008.12.006
107. Hagstrom KA, Holmes VF, Cozzarelli NR, Meyer BJ. C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes & development. 2002;16:729-42 doi:10.1101/gad.968302
108. Chu DS, Dawes HE, Lieb JD, Chan RC, Kuo AF, Meyer BJ. A molecular link between gene-specific and chromosome-wide transcriptional repression. Genes & development. 2002;16:796-805 doi:10.1101/gad.972702
109. Yonker SA, Meyer BJ. Recruitment of C. elegans dosage compensation proteins for gene-specific versus chromosome-wide repression. Development. 2003;130:6519-32 doi:10.1242/dev.00886
110. Rowland SA. Case report: ten year follow-up of lipofibroma of the median nerve in the palm. The Journal of hand surgery. 1977;2:316-7
111. Lieb JD, Albrecht MR, Chuang PT, Meyer BJ. MIX-1: an essential component of the C. elegans mitotic machinery executes X chromosome dosage compensation. Cell. 1998;92:265-77
112. Pferdehirt RR, Kruesi WS, Meyer BJ. An MLL/COMPASS subunit functions in the C. elegans dosage compensation complex to target X chromosomes for transcriptional regulation of gene expression. Genes & development. 2011;25:499-515 doi:10.1101/gad.2016011
113. Dawes HE, Berlin DS, Lapidus DM, Nusbaum C, Davis TL, Meyer BJ. Dosage compensation proteins targeted to X chromosomes by a determinant of hermaphrodite fate. Science. 1999;284:1800-4
114. Csankovszki G, McDonel P, Meyer BJ. Recruitment and spreading of the C. elegans dosage compensation complex along X chromosomes. Science. 2004;303:1182-5 doi:10.1126/science.1092938
115. Ercan S, Giresi PG, Whittle CM, Zhang X, Green RD, Lieb JD. X chromosome repression by localization of the C. elegans dosage compensation machinery to sites of transcription initiation. Nature genetics. 2007;39:403-8 doi:10.1038/ng1983
116. Ercan S, Dick LL, Lieb JD. The C. elegans dosage compensation complex propagates dynamically and independently of X chromosome sequence. Current biology: CB. 2009;19:1777-87 doi:10.1016/j.cub.2009.09.047
117. Jans J, Gladden JM, Ralston EJ, Pickle CS, Michel AH, Pferdehirt RR. et al. A condensin-like dosage compensation complex acts at a distance to control expression throughout the genome. Genes & development. 2009;23:602-18 doi:10.1101/gad.1751109
118. McDonel P, Jans J, Peterson BK, Meyer BJ. Clustered DNA motifs mark X chromosomes for repression by a dosage compensation complex. Nature. 2006;444:614-8 doi:10.1038/nature05338
119. Liu X, Lee CK, Granek JA, Clarke ND, Lieb JD. Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection. Genome research. 2006;16:1517-28 doi:10.1101/gr.5655606
120. He HH, Meyer CA, Hu SS, Chen MW, Zang C, Liu Y. et al. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nature methods. 2014;11:73-8 doi:10.1038/nmeth.2762
121. Vierstra J, Wang H, John S, Sandstrom R, Stamatoyannopoulos JA. Coupling transcription factor occupancy to nucleosome architecture with DNase-FLASH. Nature methods. 2014;11:66-72 doi:10.1038/nmeth.2713
122. He X, Chatterjee R, John S, Bravo H, Sathyanarayana BK, Biddie SC. et al. Contribution of nucleosome binding preferences and co-occurring DNA sequences to transcription factor binding. BMC genomics. 2013;14:428. doi:10.1186/1471-2164-14-428
123. van Bakel H, Tsui K, Gebbia M, Mnaimneh S, Hughes TR, Nislow C. A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS genetics. 2013;9:e1003479. doi:10.1371/journal.pgen.1003479
124. Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes & development. 2011;25:2227-41 doi:10.1101/gad.176826.111
125. Alekseyenko AA, Ho JW, Peng S, Gelbart M, Tolstorukov MY, Plachetka A. et al. Sequence-specific targeting of dosage compensation in Drosophila favors an active chromatin context. PLoS genetics. 2012;8:e1002646. doi:10.1371/journal.pgen.1002646
126. Talbert PB, Henikoff S. Spreading of silent chromatin: inaction at a distance. Nature reviews Genetics. 2006;7:793-803 doi:10.1038/nrg1920
127. Pinter SF, Sadreyev RI, Yildirim E, Jeon Y, Ohsumi TK, Borowsky M. et al. Spreading of X chromosome inactivation via a hierarchy of defined Polycomb stations. Genome research. 2012;22:1864-76 doi:10.1101/gr.133751.111
128. Simon MD, Pinter SF, Fang R, Sarma K, Rutenberg-Schoenberg M, Bowman SK. et al. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature. 2013;504:465-9 doi:10.1038/nature12719
129. Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C. et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341:1237973. doi:10.1126/science.1237973
130. Popova BC, Tada T, Takagi N, Brockdorff N, Nesterova TB. Attenuated spread of X-inactivation in an X;autosome translocation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7706-11 doi:10.1073/pnas.0602021103
131. Tang YA, Huntley D, Montana G, Cerase A, Nesterova TB, Brockdorff N. Efficiency of Xist-mediated silencing on autosomes is linked to chromosomal domain organisation. Epigenetics & chromatin. 2010;3:10. doi:10.1186/1756-8935-3-10
132. Splinter E, de Wit E, Nora EP, Klous P, van de Werken HJ, Zhu Y. et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes & development. 2011;25:1371-83 doi:10.1101/gad.633311
133. Gorchakov AA, Alekseyenko AA, Kharchenko P, Park PJ, Kuroda MI. Long-range spreading of dosage compensation in Drosophila captures transcribed autosomal genes inserted on X. Genes & development. 2009;23:2266-71 doi:10.1101/gad.1840409
134. Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, Yang P. et al. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Molecular cell. 2007;28:121-33 doi:10.1016/j.molcel.2007.08.011
135. Sural TH, Peng S, Li B, Workman JL, Park PJ, Kuroda MI. The MSL3 chromodomain directs a key targeting step for dosage compensation of the Drosophila melanogaster X chromosome. Nature structural & molecular biology. 2008;15:1318-25 doi:10.1038/nsmb.1520
136. Straub T, Zabel A, Gilfillan GD, Feller C, Becker PB. Different chromatin interfaces of the Drosophila dosage compensation complex revealed by high-shear ChIP-seq. Genome research. 2013;23:473-85 doi:10.1101/gr.146407.112
137. Kranz AL, Jiao CY, Winterkorn LH, Albritton SE, Kramer M, Ercan S. Genome-wide analysis of condensin binding in Caenorhabditis elegans. Genome biology. 2013;14:R112. doi:10.1186/gb-2013-14-10-r112
138. D'Ambrosio C, Schmidt CK, Katou Y, Kelly G, Itoh T, Shirahige K. et al. Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes & development. 2008;22:2215-27 doi:10.1101/gad.1675708
139. Lengronne A, Katou Y, Mori S, Yokobayashi S, Kelly GP, Itoh T. et al. Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature. 2004;430:573-8 doi:10.1038/nature02742
140. Glynn EF, Megee PC, Yu HG, Mistrot C, Unal E, Koshland DE. et al. Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoS biology. 2004;2:E259. doi:10.1371/journal.pbio.0020259
141. Kim JH, Zhang T, Wong NC, Davidson N, Maksimovic J, Oshlack A. et al. Condensin I associates with structural and gene regulatory regions in vertebrate chromosomes. Nature communications. 2013;4:2537. doi:10.1038/ncomms3537
142. Van Bortle K, Nichols MH, Li L, Ong CT, Takenaka N, Qin ZS. et al. Insulator function and topological domain border strength scale with architectural protein occupancy. Genome biology. 2014;15:R82. doi:10.1186/gb-2014-15-5-r82
143. Dowen JM, Bilodeau S, Orlando DA, Hubner MR, Abraham BJ, Spector DL. et al. Multiple structural maintenance of chromosome complexes at transcriptional regulatory elements. Stem cell reports. 2013;1:371-8 doi:10.1016/j.stemcr.2013.09.002
144. Tada K, Susumu H, Sakuno T, Watanabe Y. Condensin association with histone H2A shapes mitotic chromosomes. Nature. 2011;474:477-83 doi:10.1038/nature10179
145. Petty EL, Collette KS, Cohen AJ, Snyder MJ, Csankovszki G. Restricting dosage compensation complex binding to the X chromosomes by H2A.Z/HTZ-1. PLoS genetics. 2009;5:e1000699. doi:10.1371/journal.pgen.1000699
146. Kimura K, Hirano T. Dual roles of the 11S regulatory subcomplex in condensin functions. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11972-7 doi:10.1073/pnas.220326097
147. Piazza I, Rutkowska A, Ori A, Walczak M, Metz J, Pelechano V. et al. Association of condensin with chromosomes depends on DNA binding by its HEAT-repeat subunits. Nature structural & molecular biology. 2014;21:560-8 doi:10.1038/nsmb.2831
148. Wutz A. Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nature reviews Genetics. 2011;12:542-53 doi:10.1038/nrg3035
149. Brown CJ, Willard HF. The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature. 1994;368:154-6 doi:10.1038/368154a0
150. Csankovszki G, Panning B, Bates B, Pehrson JR, Jaenisch R. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nature genetics. 1999;22:323-4 doi:10.1038/11887
151. Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750-6 doi:10.1126/science.1163045
152. Cerase A, Smeets D, Tang YA, Gdula M, Kraus F, Spivakov M. et al. Spatial separation of Xist RNA and polycomb proteins revealed by superresolution microscopy. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:2235-40 doi:10.1073/pnas.1312951111
153. Wang J, Mager J, Chen Y, Schneider E, Cross JC, Nagy A. et al. Imprinted X inactivation maintained by a mouse Polycomb group gene. Nature genetics. 2001;28:371-5 doi:10.1038/ng574
154. Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H. et al. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131-5 doi:10.1126/science.1084274
155. Nozawa RS, Nagao K, Igami KT, Shibata S, Shirai N, Nozaki N. et al. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nature structural & molecular biology. 2013;20:566-73 doi:10.1038/nsmb.2532
156. Yang F, Babak T, Shendure J, Disteche CM. Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome research. 2010;20:614-22 doi:10.1101/gr.103200.109
157. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400-4 doi:10.1038/nature03479
158. Deng X, Berletch JB, Nguyen DK, Disteche CM. X chromosome regulation: diverse patterns in development, tissues and disease. Nature reviews Genetics. 2014;15:367-78 doi:10.1038/nrg3687
159. Chaumeil J, Le Baccon P, Wutz A, Heard E. A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes & development. 2006;20:2223-37 doi:10.1101/gad.380906
160. Calabrese JM, Sun W, Song L, Mugford JW, Williams L, Yee D. et al. Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell. 2012;151:951-63 doi:10.1016/j.cell.2012.10.037
161. Akhtar A, Becker PB. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Molecular cell. 2000;5:367-75
162. Smith ER, Allis CD, Lucchesi JC. Linking global histone acetylation to the transcription enhancement of X-chromosomal genes in Drosophila males. The Journal of biological chemistry. 2001;276:31483-6 doi:10.1074/jbc.C100351200
163. Kind J, Vaquerizas JM, Gebhardt P, Gentzel M, Luscombe NM, Bertone P. et al. Genome-wide analysis reveals MOF as a key regulator of dosage compensation and gene expression in Drosophila. Cell. 2008;133:813-28 doi:10.1016/j.cell.2008.04.036
164. Gelbart ME, Larschan E, Peng S, Park PJ, Kuroda MI. Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nature structural & molecular biology. 2009;16:825-32 doi:10.1038/nsmb.1644
165. Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI. High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes & development. 2006;20:848-57 doi:10.1101/gad.1400206
166. Gilfillan GD, Straub T, de Wit E, Greil F, Lamm R, van Steensel B. et al. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes & development. 2006;20:858-70 doi:10.1101/gad.1399406
167. Allahverdi A, Yang R, Korolev N, Fan Y, Davey CA, Liu CF. et al. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic acids research. 2011;39:1680-91 doi:10.1093/nar/gkq900
168. Liu Y, Lu C, Yang Y, Fan Y, Yang R, Liu CF. et al. Influence of histone tails and H4 tail acetylations on nucleosome-nucleosome interactions. Journal of molecular biology. 2011;414:749-64 doi:10.1016/j.jmb.2011.10.031
169. Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ. et al. X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature. 2011;471:115-8 doi:10.1038/nature09757
170. Regnard C, Straub T, Mitterweger A, Dahlsveen IK, Fabian V, Becker PB. Global analysis of the relationship between JIL-1 kinase and transcription. PLoS genetics. 2011;7:e1001327. doi:10.1371/journal.pgen.1001327
171. Ferrari F, Plachetka A, Alekseyenko AA, Jung YL, Ozsolak F, Kharchenko PV. et al. "Jump start and gain" model for dosage compensation in Drosophila based on direct sequencing of nascent transcripts. Cell reports. 2013;5:629-36 doi:10.1016/j.celrep.2013.09.037
172. Prabhakaran M, Kelley RL. Mutations in the transcription elongation factor SPT5 disrupt a reporter for dosage compensation in Drosophila. PLoS genetics. 2012;8:e1003073. doi:10.1371/journal.pgen.1003073
173. Conrad T, Cavalli FM, Vaquerizas JM, Luscombe NM, Akhtar A. Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters. Science. 2012;337:742-6 doi:10.1126/science.1221428
174. Dunlap D, Yokoyama R, Ling H, Sun HY, McGill K, Cugusi S. et al. Distinct contributions of MSL complex subunits to the transcriptional enhancement responsible for dosage compensation in Drosophila. Nucleic acids research. 2012;40:11281-91 doi:10.1093/nar/gks890
175. Cugusi S, Ramos E, Ling H, Yokoyama R, Luk KM, Lucchesi JC. Topoisomerase II plays a role in dosage compensation in Drosophila. Transcription. 2013;4:238-50
176. Furuhashi H, Nakajima M, Hirose S. DNA supercoiling factor contributes to dosage compensation in Drosophila. Development. 2006;133:4475-83 doi:10.1242/dev.02620
177. Grimaud C, Becker PB. The dosage compensation complex shapes the conformation of the X chromosome in Drosophila. Genes & development. 2009;23:2490-5 doi:10.1101/gad.539509
178. Mendjan S, Taipale M, Kind J, Holz H, Gebhardt P, Schelder M. et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Molecular cell. 2006;21:811-23 doi:10.1016/j.molcel.2006.02.007
179. Vielle A, Lang J, Dong Y, Ercan S, Kotwaliwale C, Rechtsteiner A. et al. H4K20me1 contributes to downregulation of X-linked genes for C. elegans dosage compensation. PLoS genetics. 2012;8:e1002933. doi:10.1371/journal.pgen.1002933
180. Liu T, Rechtsteiner A, Egelhofer TA, Vielle A, Latorre I, Cheung MS. et al. Broad chromosomal domains of histone modification patterns in C. elegans. Genome research. 2011;21:227-36 doi:10.1101/gr.115519.110
181. Wells MB, Snyder MJ, Custer LM, Csankovszki G. Caenorhabditis elegans dosage compensation regulates histone H4 chromatin state on X chromosomes. Molecular and cellular biology. 2012;32:1710-9 doi:10.1128/MCB.06546-11
182. Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y. et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Molecular cell. 2002;9:1201-13
183. Serrano L, Martinez-Redondo P, Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P. et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes & development. 2013;27:639-53 doi:10.1101/gad.211342.112
184. Kapoor-Vazirani P, Vertino PM. A dual role for the histone methyltransferase PR-SET7/SETD8 and histone H4 lysine 20 monomethylation in the local regulation of RNA polymerase II pausing. The Journal of biological chemistry. 2014;289:7425-37 doi:10.1074/jbc.M113.520783
185. Beck DB, Oda H, Shen SS, Reinberg D. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes & development. 2012;26:325-37 doi:10.1101/gad.177444.111
186. Oda H, Okamoto I, Murphy N, Chu J, Price SM, Shen MM. et al. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Molecular and cellular biology. 2009;29:2278-95 doi:10.1128/MCB.01768-08
187. Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL. et al. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Molecular cell. 2010;40:364-76 doi:10.1016/j.molcel.2010.10.011
188. Rice JC, Nishioka K, Sarma K, Steward R, Reinberg D, Allis CD. Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes & development. 2002;16:2225-30 doi:10.1101/gad.1014902
189. Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT. et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508-12 doi:10.1038/nature09272
190. Lim HJ, Dimova NV, Tan MK, Sigoillot FD, King RW, Shi Y. The G2/M regulator histone demethylase PHF8 is targeted for degradation by the anaphase-promoting complex containing CDC20. Molecular and cellular biology. 2013;33:4166-80 doi:10.1128/MCB.00689-13
191. Wilkins BJ, Rall NA, Ostwal Y, Kruitwagen T, Hiragami-Hamada K, Winkler M. et al. A cascade of histone modifications induces chromatin condensation in mitosis. Science. 2014;343:77-80 doi:10.1126/science.1244508
192. Ercan S, Lubling Y, Segal E, Lieb JD. High nucleosome occupancy is encoded at X-linked gene promoters in C. elegans. Genome research. 2011;21:237-44 doi:10.1101/gr.115931.110
193. Zhang Y, Malone JH, Powell SK, Periwal V, Spana E, Macalpine DM. et al. Expression in aneuploid Drosophila S2 cells. PLoS biology. 2010;8:e1000320. doi:10.1371/journal.pbio.1000320
194. Strome S, Kelly WG, Ercan S, Lieb JD. Regulation of the X chromosomes in Caenorhabditis elegans. Cold Spring Harbor perspectives in biology. 2014 6. doi:10.1101/cshperspect.a018366
195. Wallis MC, Waters PD, Graves JA. Sex determination in mammals--before and after the evolution of SRY. Cellular and molecular life sciences: CMLS. 2008;65:3182-95 doi:10.1007/s00018-008-8109-z
196. Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ. et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990;346:240-4 doi:10.1038/346240a0
197. Berta P, Hawkins JR, Sinclair AH, Taylor A, Griffiths BL, Goodfellow PN. et al. Genetic evidence equating SRY and the testis-determining factor. Nature. 1990;348:448-50 doi:10.1038/348448A0
198. Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R. Male development of chromosomally female mice transgenic for Sry. Nature. 1991;351:117-21 doi:10.1038/351117a0
199. Salz HK. Sex determination in insects: a binary decision based on alternative splicing. Current opinion in genetics & development. 2011;21:395-400 doi:10.1016/j.gde.2011.03.001
200. Bashaw GJ, Baker BS. The regulation of the Drosophila msl-2 gene reveals a function for Sex-lethal in translational control. Cell. 1997;89:789-98
201. Kelley RL, Wang J, Bell L, Kuroda MI. Sex lethal controls dosage compensation in Drosophila by a non-splicing mechanism. Nature. 1997;387:195-9 doi:10.1038/387195a0
202. Gebauer F, Merendino L, Hentze MW, Valcarcel J. The Drosophila splicing regulator sex-lethal directly inhibits translation of male-specific-lethal 2 mRNA. RNA. 1998;4:142-50
203. Graindorge A, Carre C, Gebauer F. Sex-lethal promotes nuclear retention of msl2 mRNA via interactions with the STAR protein HOW. Genes & development. 2013;27:1421-33 doi:10.1101/gad.214999.113
204. Miller LM, Plenefisch JD, Casson LP, Meyer BJ. xol-1: a gene that controls the male modes of both sex determination and X chromosome dosage compensation in C. elegans. Cell. 1988;55:167-83
205. Rhind NR, Miller LM, Kopczynski JB, Meyer BJ. xol-1 acts as an early switch in the C. elegans male/hermaphrodite decision. Cell. 1995;80:71-82
206. Gladden JM, Farboud B, Meyer BJ. Revisiting the X:A signal that specifies Caenorhabditis elegans sexual fate. Genetics. 2007;177:1639-54 doi:10.1534/genetics.107.078071
207. Powell JR, Jow MM, Meyer BJ. The T-box transcription factor SEA-1 is an autosomal element of the X:A signal that determines C. elegans sex. Developmental cell. 2005;9:339-49 doi:10.1016/j.devcel.2005.06.009
208. Carmi I, Kopczynski JB, Meyer BJ. The nuclear hormone receptor SEX-1 is an X-chromosome signal that determines nematode sex. Nature. 1998;396:168-73 doi:10.1038/24164
209. Farboud B, Nix P, Jow MM, Gladden JM, Meyer BJ. Molecular antagonism between X-chromosome and autosome signals determines nematode sex. Genes & development. 2013;27:1159-78 doi:10.1101/gad.217026.113
210. Nicoll M, Akerib CC, Meyer BJ. X-chromosome-counting mechanisms that determine nematode sex. Nature. 1997;388:200-4 doi:10.1038/40669
211. Takagi N, Abe K. Detrimental effects of two active X chromosomes on early mouse development. Development. 1990;109:189-201
212. Belote JM, Lucchesi JC. Male-specific lethal mutations of Drosophila melanogaster. Genetics. 1980;96:165-86
213. Davis TL, Meyer BJ. SDC-3 coordinates the assembly of a dosage compensation complex on the nematode X chromosome. Development. 1997;124:1019-31
214. Villeneuve AM, Meyer BJ. The role of sdc-1 in the sex determination and dosage compensation decisions in Caenorhabditis elegans. Genetics. 1990;124:91-114
215. Plenefisch JD, DeLong L, Meyer BJ. Genes that implement the hermaphrodite mode of dosage compensation in Caenorhabditis elegans. Genetics. 1989;121:57-76
216. Custer LM, Snyder MJ, Flegel K, Csankovszki G. The onset of C. elegans dosage compensation is linked to the loss of developmental plasticity. Developmental biology. 2014;385:279-90 doi:10.1016/j.ydbio.2013.11.001
217. Minkovsky A, Patel S, Plath K. Concise review: Pluripotency and the transcriptional inactivation of the female Mammalian X chromosome. Stem Cells. 2012;30:48-54 doi:10.1002/stem.755
218. Morey R, Laurent LC. Getting off the ground state: X chromosome inactivation knocks down barriers to differentiation. Cell stem cell. 2014;14:131-2 doi:10.1016/j.stem.2014.01.012
219. Schulz EG, Meisig J, Nakamura T, Okamoto I, Sieber A, Picard C. et al. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell stem cell. 2014;14:203-16 doi:10.1016/j.stem.2013.11.022
Author contact
![]() Corresponding author: Sevinç Ercan, email: se71edu, phone: 212-992-9518.
Corresponding author: Sevinç Ercan, email: se71edu, phone: 212-992-9518.