ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2015; 3:75-81. doi:10.7150/jgen.12863 This volume Cite
Research Paper
GLAD: an Online Database of G ene L ist A nnotation for D rosophila
Yanhui Hu1 ![]() , Aram Comjean1, Lizabeth A. Perkins1, Norbert Perrimon1,2, Stephanie E. Mohr1
, Aram Comjean1, Lizabeth A. Perkins1, Norbert Perrimon1,2, Stephanie E. Mohr1
1. Drosophila RNAi Screening Center, Department of Genetics, Harvard Medical School, 77 Avenue Louis Pasteur, Boston, MA 02115, USA
2. Howard Hughes Medical Institute, 77 Avenue Louis Pasteur, Boston, MA 02115, USA
Published 2015-7-1
Abstract
We present a resource of high quality lists of functionally related Drosophila genes, e.g. based on protein domains (kinases, transcription factors, etc.) or cellular function (e.g. autophagy, signal transduction). To establish these lists, we relied on different inputs, including curation from databases or the literature and mapping from other species. Moreover, as an added curation and quality control step, we asked experts in relevant fields to review many of the lists. The resource is available online for scientists to search and view, and is editable based on community input. Annotation of gene groups is an ongoing effort and scientific need will typically drive decisions regarding which gene lists to pursue. We anticipate that the number of lists will increase over time; that the composition of some lists will grow and/or change over time as new information becomes available; and that the lists will benefit the scientific community, e.g. at experimental design and data analysis stages. Based on this, we present an easily updatable online database, available at
Keywords: GLAD, Drosophila, genes
Introduction
The Drosophila genome was first published in 2000 [1, 2] and so far five major updates to the genome assembly have been released (versions 2-6) [3, 4]. Based on recent FlyBase release (FB2015_02, May 4, 2015), the Drosophila genome is thought to contain 17,622 annotated genes, of which 13,903 are protein-coding genes. There are many advantages to interrogate the full Drosophila genome, such as in genetic or RNAi screens. However, often it is more appropriate and/or more feasible to screen a sub-set of genes, such as due to limitations on time, availability (e.g. of reagents) and/or costs. Choosing an appropriate sub-set of genes is largely guided by scientific interest. In some cases, researchers build lists for functional studies based on other 'omics data, such as transcriptomics or proteomics data, prior to a functional genomics study. In other cases, genes are grouped based on common features such as biochemical functions (e.g. kinases) or biological processes. In either case, the quality and completeness of the library will impact results, as genes inadvertently left off a list will not be included in the study, and the presence of genes that do not belong on the list will needlessly use up resources and/or affect the analysis of the results.
The availability of high quality annotated groups of related genes (hereafter, “gene groups”) allow scientists to quickly focus on relevant genes, such as in the context of functional genomics screens in tissue culture cells or in vivo. However, mechanisms for distribution and update of gene groups have been limited. At the time the Drosophila genome was published, efforts were made to compile several gene groups (see for example [5-8]). However, given the number of changes to gene annotations since then, both in terms of defining genes and understanding their functions, updating and adding to existing lists becomes important. Over the past years, the Drosophila RNAi Screening Center (DRSC) at Harvard Medical School (HMS) has put together several gene groups based on the needs of specific screening projects, as well as to support organization of reagent collections at the Transgenic RNAi Project (TRiP) at HMS. We have recognized over time a) that gene groups are of value to the community for applications additional to RNAi screens; b) that the gene groups benefit from careful curation and review by experts; and c) that the lists change over time, such that they would benefit from being available in an easily updateable database rather than as static lists. We expect gene groups to be of value to researchers at the study design stage, where the lists can help guide decisions regarding what genes are interrogated in a given screen or other assay, and at data analysis stages, such as by providing a supplement to existing groups used in gene set enrichment analyses.
Results & Discussion
Compilation and annotation strategy for gene groups
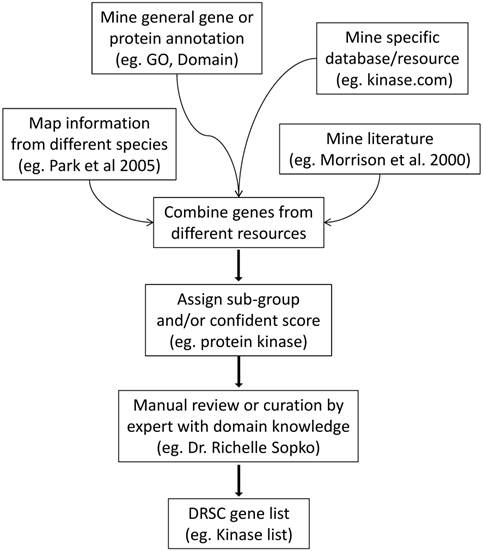
As mentioned above, several gene groups were annotated in conjunction with release of the Drosophila genome in 2000 (see for example [5-8]). More recently, FlyBase has begun to associate a number of genes with gene groups. The first release of the FlyBase gene groups (FB2015_02, released May 4, 2015) includes 178 gene groups, with the number of genes in a group ranging from 1 to 168. As we had high-throughput functional genomics screening in mind, the approaches we took to defining, building and annotating gene groups draw on knowledge not available in 2000 and are complementary to the approaches taken by FlyBase. In general, our focus is on larger sets of genes and, given the goals of large-scale functional genomics, we tend to cast a broader net, applying less stringent cut-offs for inclusion in a gene group. So far, we have annotated 23 major gene groups with 29 sub-groups. For example, kinases are annotated as belonging to one of two subgroups: protein kinases and non-protein kinases, and the transcription factors (TFs), related proteins and other DNA-binding proteins are organized into four groups, DNA-binding with transcription factor activity; transcriptional co-factors; chromatin regulation; and possible TFs, which we assign to proteins predicted to be TF based only on low confidence data (Table 1). The number of genes in major gene groups ranges from 53 to 3,683. Currently, GO annotation is the major resource for identification of groups of genes relevant to a particular molecular function, biological process or sub-cellular localization. Several individual laboratories have also built databases in particular areas (e.g. GlycoFly [9] and FlyTF [10]). In addition, a large amount of information exists in free text format in the literature. Although our strategy differed for each group, depending on available resources, in general we built the lists using one or more of the following approaches: a) mining of organized and digitalized information from existing annotation resources and databases including generic gene and protein annotation, e.g. gene ontology and UniProt, as well as specialized resources, e.g. transporterDB [11] or FlyTF [10, 12]; b) mining of information in free text format from the literature; c) mining lists from relevant publications on Drosophila or other species; d) direct curation or review by experts (Table 1). The strategy used to build a Drosophila kinases gene group is outlined in Fig. 1. To help guide studies or analyses that use the gene groups, when possible we have assigned confidence scores that help separate high- and low-confidence associations of a gene with a given group. See Methods and Table 1 for additional details regarding annotation.
Summary of approaches to gene group compilation, annotation and expert review.
| Gene group | Sub-group | Source |
|---|---|---|
| Autophagy-related* | Mapped from literature [17] | |
| Chaperone and heat shock proteins | GO/UniProt annotation supplemented by human chaperon/HSP list from Dr. Susan Lindquist lab | |
| Cytoskeletal | Interactive Fly, UniProt and publication [6], reviewed by Dr. Norbert Perrimon | |
| Glycoproteins | GlycoFly database [9] | |
| GPCRs* | GO annotation, in collaboration with Dr. Mathias Beller | |
| Kinases* | Non-protein Kinase | Nomenclature/GO/domain annotation supplemented with human KP list [23] and publications [22] [7], reviewed by Dr. Richelle Sopko |
| Protein Kinase | ||
| Phosphatases* | Non-protein Phosphatase | |
| Protein Phosphatase | ||
| Major signaling pathways | Imd pathway | flyReactome, reviewed by Dr. Herve Agaisse |
| Toll pathway | flyReactome, reviewed by Dr. Herve Agaisse | |
| Planar Cell Polarity pathway | flyReactome, reviewed by Dr. Jeff Axelrod | |
| Circadian Clock pathway | flyReactome, reviewed by Dr. Phillip Karpowicz | |
| EGFR and PVR RTK signaling pathway | Manually assembled by Dr. Norbert Perrimon | |
| FGFR signaling pathway | ||
| HEDGEHOG signaling pathway | ||
| HIPPO signaling pathway | ||
| INSULIN signaling pathway | ||
| JAK/STAT signaling pathway | ||
| NOTCH signaling pathway | ||
| TGF beta signaling pathway | ||
| TNF alpha signaling pathway | ||
| WNT signaling pathway | ||
| Nuclear hormone receptor | SignaLink [24], reviewed by Dr. Henry Krause | |
| Metabolic | Enzyme | KEGG,UniProt and mapped from publication [25] |
| Other | ||
| Mitochondrial | GO/UniProt/Mito databases [26],[27] and publications [18][19][20][28]) | |
| Nuclear-encoded oxidative phosphorylation | MitoComp2 database [29] | |
| Peroxisomal | Publication [30], UniProt/GO | |
| Proteasome | GO annotation, reviewed by Dr. Jonathan Zirin | |
| Receptors | GO/UniProt/GPCR/mapped from Human Receptome [31] | |
| Ribosome | GO annotation, reviewed by Dr. Ralph Neumuller | |
| RNA-binding* | GO/UniProt/InterPro, in collaboration with Dr. Bing Ye | |
| Secreted proteins | UniProt annotation, reviewed by Dr. Norbert Perrimon | |
| Serine proteases | UniProt annotation, reviewed by Dr. Norbert Perrimon | |
| Spliceosome | GO/UniProt/Publication [32] | |
| Transcription factors, related proteins and other DNA-binding proteins* | DNA-binding with transcription factor activity | GO/domain annotation and FlyTF.org [10] |
| Co-factor | ||
| Chromatin regulation | ||
| Maybe TF | ||
| Trans-membrane proteins* | DRSC in collaboration with NYU supplemented by TMHMM prediction [33] | |
| Transporters | ATP-Dependent | TransporterDB [11] |
| Ion Channels | ||
| Secondary Transporter | ||
| Unclassified | ||
| Ubiquitin-related* | DRSC in collaboration with NYU |
* Indicates availability of a corresponding DRSC RNAi library for cell-based screens.
Strategy for assembly of a Drosophila kinases gene group. As outlined, we incorporated information from several sources. See main text and Table 1 for relevant URLs and reference citations.
Features of the user interface
The gene group resource, which we call GLAD for gene list annotation for Drosophila, is available online at www.flyrnai.org/glad. Users can choose a gene group of interest from a drop-down menu. At the results page (Fig. 2), information about how the list was built is indicated, along with detailed information regarding members of the group. The table of genes includes FlyBase gene identifiers, gene symbols, sub-group annotations and if available, a confidence score. Tables can be downloaded for off-line analysis. At the user interface, a link to UP-TORR [13] is provided so users can quickly identify corresponding cell-based or in vivo RNAi reagents from public resources. A form provides an opportunity for the research community at large to suggest changes or additions to a given list (see below).
User interface for the GLAD gene group resource (www.flyrnai.org/glad). Features of the user interface include search or download of specific lists, one-click transfer of the list to UP-TORR for identification of corresponding RNAi reagents, and the option to provide feedback regarding a list (e.g. suggest new genes or relevant publications).
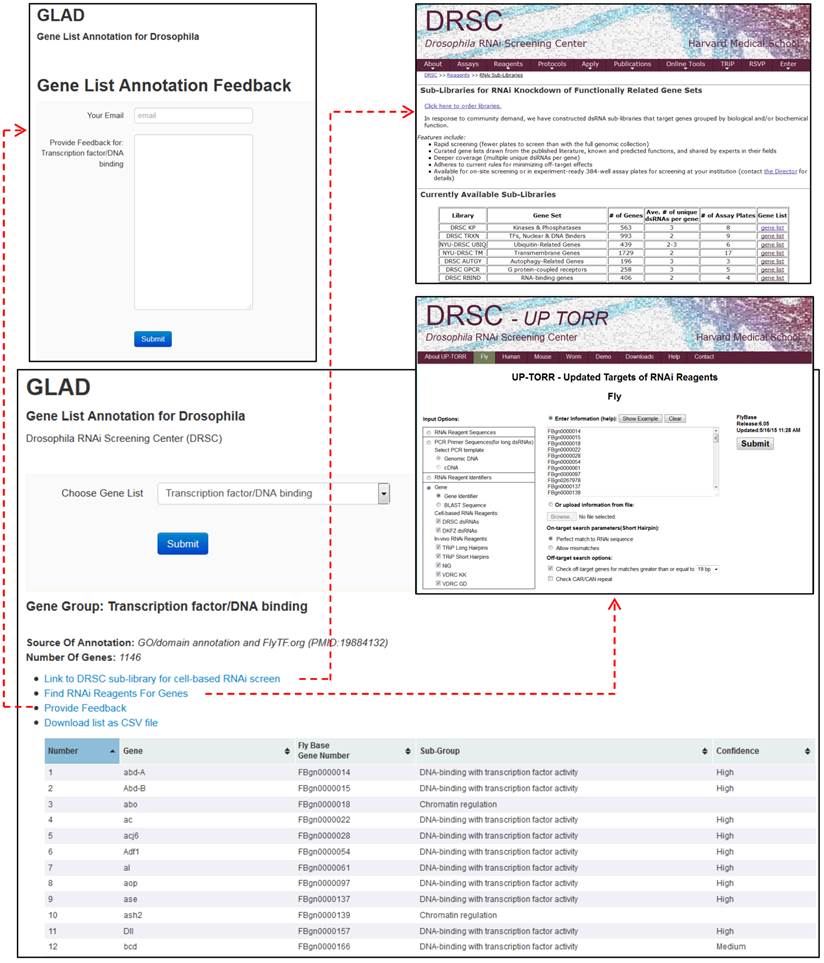
Feedback will improve the quality of the resource
Although we have made a concerted effort to evaluate all available resources and used best available methods for building each list, there remains room for improvement, in particular as new knowledge places new genes in a given group. To facilitate community updates, we welcome and encourage researchers to use the form at each gene group list to provide feedback and/or alert us to relevant publications. We will evaluate feedback and modify gene groups accordingly. Annotation of new gene groups is also an ongoing effort, and scientific interest will typically drive decisions regarding which gene groups to build next. We welcome feedback from the community regarding which groups not already covered by GLAD or FlyBase gene groups should be added. With community input as well as continued curation by bioinformatics experts, we anticipate that the GLAD resource will improve and expand over time, further increasing its value to the community. Examples of studies that used these gene groups include a primary cell-based screen of autophagy-related factors [14] and an in vivo screen of transcription factors [15].
Methods
Compilation of gene groups
Specific resources used to annotate the gene groups are shown in Table 1. Five groups listed below exemplify the range of approaches we took. 1) Most of the major signal transduction pathways were assembled manually by Dr. N. Perrimon. 2) The main source for the autophagy-related factors list was orthologs [16] of factors identified in a mammalian proteomics study [17]. 3) The mitochondrial gene group was built by combining gene annotation, relevant databases (MitoDrome, MitoMiner), mapping of mammalian orthologs and experimental proteomics data [18-20]. 4) The transcription factors and other DNA-binding factors list was built based on GO annotation and domain annotation. More specifically, the genes annotated with relevant GO terms such as “sequence-specific DNA binding transcription factor activity” and/or genes annotated with relevant domain at InterPro database. The list was then supplemented with the genes annotated at FlyTF database [10, 12]. The sub-categories were assigned based on GO term and high confidence was assigned to TFs associated with experimental evidence at FlyTF database. 5) As outlined in Fig. 1, the kinase list was initially assembled based on genes annotated as having “kinase activity,” supplemented by genes annotated at InterPro [21] as containing a “kinase” domain, supplemented with information from two publications [7, 22], and further supplemented following mapping of Drosophila orthologs of human genes included on a list of kinases [23]. The compiled list was reviewed by Dr. Richelle Sopko with specific expertise and interest in the field. Family assignment was applied according to Manning et al. [22].
User interface implementation
The GLAD website was created at the DRSC and hosted on web servers provided and maintained by the Harvard Medical School (HMS) Research Computing Group. The website was created in PHP using silex as a web framework. The PHP web code pulls the lists from tables from the mysql database of flyrnai and prepares data for display. HTML/javascript and jquery was used to create sortable output tables.
Acknowledgements
We would like to thank Drs. Herve Agaisse (Yale School of Medicine), Jeff Axelrod (Stanford School of Medicine), Mathias Beller (Heinrich Heine Universität Düsseldorf), Phillip Karpowicz (University of Winsor), John J. Kim (University of Michigan), Henry Krause (University of Toronto), Susan Lindquist (MIT), Ralph Neumuller (Harvard Medical School), Richelle Sopko (Harvard Medical School), Bing Ye (University of Michigan) and Jonathan Zirin (Harvard Medical School) for taking the time to review specific gene groups. We also thank Dr. Steven Marygold at FlyBase for helpful discussion. The DRSC and TRiP are supported by NIH NIGMS R01 GM067761, R01 GM084947 and R24 RR032668. SEM is additionally supported in part by the Dana Farber/Harvard Cancer Center, which is supported in part by NCI Cancer Center Support Grant # NIH 5 P30 CA06516. NP is an Investigator with the Howard Hughes Medical Institute.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG. et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185-95
2. Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ. et al. A whole-genome assembly of Drosophila. Science. 2000;287:2196-204
3. dos Santos G, Schroeder AJ, Goodman JL, Strelets VB, Crosby MA, Thurmond J. et al. FlyBase: introduction of the Drosophila melanogaster Release 6 reference genome assembly and large-scale migration of genome annotations. Nucleic Acids Res. 2015;43:D690-7 doi:10.1093/nar/gku1099
4. Drysdale R. FlyBase: a database for the Drosophila research community. Methods Mol Biol. 2008;420:45-59 doi:10.1007/978-1-59745-583-1_3
5. Brody T, Cravchik A. Drosophila melanogaster G protein-coupled receptors. J Cell Biol. 2000;150:F83-8
6. Goldstein LS, Gunawardena S. Flying through the drosophila cytoskeletal genome. J Cell Biol. 2000;150:F63-8
7. Morrison DK, Murakami MS, Cleghon V. Protein kinases and phosphatases in the Drosophila genome. J Cell Biol. 2000;150:F57-62
8. Lasko P. The drosophila melanogaster genome: translation factors and RNA binding proteins. J Cell Biol. 2000;150:F51-6
9. Baycin-Hizal D, Tian Y, Akan I, Jacobson E, Clark D, Chu J. et al. GlycoFly: a database of Drosophila N-linked glycoproteins identified using SPEG--MS techniques. J Proteome Res. 2011;10:2777-84 doi:10.1021/pr200004t
10. Pfreundt U, James DP, Tweedie S, Wilson D, Teichmann SA, Adryan B. FlyTF: improved annotation and enhanced functionality of the Drosophila transcription factor database. Nucleic Acids Res. 2010;38:D443-7 doi:10.1093/nar/gkp910
11. Ren Q, Chen K, Paulsen IT. TransportDB: a comprehensive database resource for cytoplasmic membrane transport systems and outer membrane channels. Nucleic Acids Res. 2007;35:D274-9 doi:10.1093/nar/gkl925
12. Adryan B, Teichmann SA. FlyTF: a systematic review of site-specific transcription factors in the fruit fly Drosophila melanogaster. Bioinformatics. 2006;22:1532-3 doi:10.1093/bioinformatics/btl143
13. Hu Y, Roesel C, Flockhart I, Perkins L, Perrimon N, Mohr SE. UP-TORR: online tool for accurate and Up-to-Date annotation of RNAi Reagents. Genetics. 2013;195:37-45 doi:10.1534/genetics.113.151340
14. Zirin J, Nieuwenhuis J, Samsonova A, Tao R, Perrimon N. Regulators of autophagosome formation in Drosophila muscles. PLoS Genet. 2015;11:e1005006. doi:10.1371/journal.pgen.1005006
15. Karpowicz P, Zhang Y, Hogenesch JB, Emery P, Perrimon N. The circadian clock gates the intestinal stem cell regenerative state. Cell reports. 2013;3:996-1004 doi:10.1016/j.celrep.2013.03.016
16. Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N. et al. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics. 2011;12:357. doi:10.1186/1471-2105-12-357
17. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68-76 doi:10.1038/nature09204
18. Lotz C, Lin AJ, Black CM, Zhang J, Lau E, Deng N. et al. Characterization, design, and function of the mitochondrial proteome: from organs to organisms. J Proteome Res. 2014;13:433-46 doi:10.1021/pr400539j
19. Yin S, Xue J, Sun H, Wen B, Wang Q, Perkins G. et al. Quantitative evaluation of the mitochondrial proteomes of Drosophila melanogaster adapted to extreme oxygen conditions. PLoS One. 2013;8:e74011. doi:10.1371/journal.pone.0074011
20. Chen C, Hu Y, Udeshi ND, Lau TY, Wirtz-Peitz F, He L. et al. Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase. Proc Natl Acad Sci U S A. 2015 submitted
21. Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M. et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001;29:37-40
22. Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. 2002;27:514-20
23. Park J, Hu Y, Murthy TV, Vannberg F, Shen B, Rolfs A. et al. Building a human kinase gene repository: bioinformatics, molecular cloning, and functional validation. Proc Natl Acad Sci U S A. 2005;102:8114-9 doi:10.1073/pnas.0503141102
24. Fazekas D, Koltai M, Turei D, Modos D, Palfy M, Dul Z. et al. SignaLink 2 - a signaling pathway resource with multi-layered regulatory networks. BMC Syst Biol. 2013;7:7. doi:10.1186/1752-0509-7-7
25. Ros S, Santos CR, Moco S, Baenke F, Kelly G, Howell M. et al. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer discovery. 2012;2:328-43 doi:10.1158/2159-8290.CD-11-0234
26. Sardiello M, Licciulli F, Catalano D, Attimonelli M, Caggese C. MitoDrome: a database of Drosophila melanogaster nuclear genes encoding proteins targeted to the mitochondrion. Nucleic Acids Res. 2003;31:322-4
27. Smith AC, Blackshaw JA, Robinson AJ. MitoMiner: a data warehouse for mitochondrial proteomics data. Nucleic Acids Res. 2012;40:D1160-7 doi:10.1093/nar/gkr1101
28. Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA. et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328-31 doi:10.1126/science.1230593
29. Porcelli D, Barsanti P, Pesole G, Caggese C. The nuclear OXPHOS genes in insecta: a common evolutionary origin, a common cis-regulatory motif, a common destiny for gene duplicates. BMC Evol Biol. 2007;7:215. doi:10.1186/1471-2148-7-215
30. Faust JE, Verma A, Peng C, McNew JA. An inventory of peroxisomal proteins and pathways in Drosophila melanogaster. Traffic. 2012;13:1378-92 doi:10.1111/j.1600-0854.2012.01393.x
31. Ben-Shlomo I, Yu Hsu S, Rauch R, Kowalski HW, Hsueh AJ. Signaling receptome: a genomic and evolutionary perspective of plasma membrane receptors involved in signal transduction. Sci STKE. 2003;2003:RE9. doi:10.1126/stke.2003.187.re9
32. Gan Q, Chepelev I, Wei G, Tarayrah L, Cui K, Zhao K. et al. Dynamic regulation of alternative splicing and chromatin structure in Drosophila gonads revealed by RNA-seq. Cell Res. 2010;20:763-83 doi:10.1038/cr.2010.64
33. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567-80 doi:10.1006/jmbi.2000.4315
Author contact
![]() Corresponding author: Yanhui Hu, yanhui_huharvard.edu
Corresponding author: Yanhui Hu, yanhui_huharvard.edu