ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2016; 4:42-47. doi:10.7150/jgen.16156 This volume Cite
Short Research Communication
Whole Genome Sequence Analysis of an Alachlor and Endosulfan Degrading Micrococcus sp. strain 2385 Isolated from Ochlockonee River, Florida
Ashish Pathak1, Ashvini Chauhan1 ![]() , Ayman Y.I. Ewida2, Paul Stothard3
, Ayman Y.I. Ewida2, Paul Stothard3
1. Environmental Biotechnology and Genomics Laboratory, School of the Environment, 1515 S. Martin Luther King Jr. Blvd., Suite 305B, FSH Science Research Center, Florida A&M University, Tallahassee, FL- 32307, USA.
2. Department of Microbiology, Central Laboratory for Environmental Quality Monitoring (CLEQM), National Water Research Center, Egypt.
3. Department of Agricultural, Food and Nutritional Science, University of Alberta, Edmonton, AB T6G2P5, Canada.
Published 2016-9-8
Abstract

We recently isolated Micrococcus sp. strain 2385 from Ochlockonee River, Florida and demonstrated potent biodegradative activity against two commonly used pesticides- alachlor [(2-chloro-2`,6`-diethylphenyl-N (methoxymethyl)acetanilide)] and endosulfan [(6,7,8,9,10,10-hexachloro-1,5,5a,6,9,9a-hexahydro-6,9methano-2,3,4-benzo(e)di-oxathiepin-3-oxide], respectively. To further identify the repertoire of metabolic functions possessed by strain 2385, a draft genome sequence was obtained, assembled, annotated and analyzed. The genome sequence of Micrococcus sp. strain 2385 consisted of 1,460,461,440 bases which assembled into 175 contigs with an N50 contig length of 50,109 bases and a coverage of 600x. The genome size of this strain was estimated at 2,431,226 base pairs with a G+C content of 72.8 and a total number of 2,268 putative genes. RAST annotated a total of 340 subsystems in the genome of strain 2385 along with the presence of 2,177 coding sequences. A genome wide survey indicated that that strain 2385 harbors a plethora of genes to degrade other pollutants including caprolactam, PAHs (such as naphthalene), styrene, toluene and several chloroaromatic compounds.
Keywords: Alachlor, Endosulfan, Biodegradation, Whole Genome Sequencing (WGS), Micrococcus.
Introduction
Estimates indicate that approximately three million metric tons of pesticides are applied each year across the globe 1 and according to the World Health Organization (WHO), each year, approximately three million people are poisoned by pesticides and herbicides [http://whqlibdoc.who.int/publications/1990/9241561394.pdf]. Endosulfan [(6,7,8,9,10,10-hexachloro-1,5,5a,6,9,9a-hexahydro-6,9methano-2,3,4-benzo(e)di-oxathiepin-3-oxide] and alachlor [(2-chloro-2`,6`-diethylphenyl-N (methoxymethyl)acetanilide)], are among those pesticides used widely to enhance crop productivity. Endosulfan is a broad-spectrum organochlorine insecticide broadly used to control agricultural pests found on a variety of plants to include fruits and vegetables. Alachlor is an acetanilide herbicide and is generally used to control grasses and broadleaf weeds on agricultural cropland, including corn, soybeans, peanuts and other crops, and on non-crop land for general weed control. Even though alachlor has low toxicity, but repeated exposure is known to cause hepato-toxicity and even tumor formation in rats 2. United States Environmental Protection Agency (USEPA) lists alachlor as a potential human carcinogen, but only at high doses (http://www.epa.gov/pesticides/).
Once pesticides are released into the environment, they can be microbially degraded if favorable conditions persist. We recently isolated a suite of bacteria with the ability to degrade both endosulfan and alachlor using standard enrichment techniques containing pesticides as the sole source of carbon and energy 3. This resulted in the isolation of several potent pesticide degrading strains among which, Micrococcus sp. strain 2385 degraded 55% and 93% of the spiked alachlor and endosulfan, respectively, over a 4-week period 3. Originally, the strain 2385 was classified as a Burkholderia sp., but corrected to Micrococcus luteus based on the whole genome sequence analysis. It has been previously shown that Micrococcus varians can be used to remediate oil-contaminated sites 4, Kerosene hydrocarbon 5 and other pollutants such as PAHs 6. Thus further research is required on the genome-wide understanding of Micrococcus spp. and their metabolic potential. Towards this end, we report our findings on the genomic analysis of Micrococcus sp. strain 2385 which demonstrates a plethora of environmentally-relevant biodegradative traits possessed by this strain.
Briefly, genomic DNA was extracted from strain 2385 and prepared for sequencing on an Illumina HiSeq2000 instrument as described previously 7. The genome was de novo assembled using paired-end reads using the software package CLC Genomics Workbench V6.0 (CLCbio, Cambridge, MA). The assembled genome, with a coverage of 600x, was then annotated and genes predicted by IMG ER 8, RAST 9 and NCBI's Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP), version 2.0. A circular genomic map of strain 2385 was obtained using CGView 10 using the 175 assembled contigs with N50 of 50,109 bases (Fig. 1). The genome size of strain 2385 was estimated to be approximately 2,431,226 bp with a G+C content of 72.8. Moreover, strain 2385 was contained a total of 2,268 putative genes, with 4 copies of the 16S, 49 trRNA genes and 64 total RNA genes, respectively.
Circular genome map of Micrococcus sp. strain 2385 with the first (outermost) and fourth rings depicting COG categories of protein coding genes on the forward and reverse strands, respectively. The second and third rings show the locations of protein coding, tRNA, and rRNA genes on the forward and reverse strands, respectively. The black plot depicts GC content with the peaks extending towards the outside of the circle representing GC content above the genome average, whereas those extending towards the center mark segments with GC content lower than the genome average. The innermost plot depicts GC skew. Both base composition plots were generated using a sliding window of 50,000 nt.

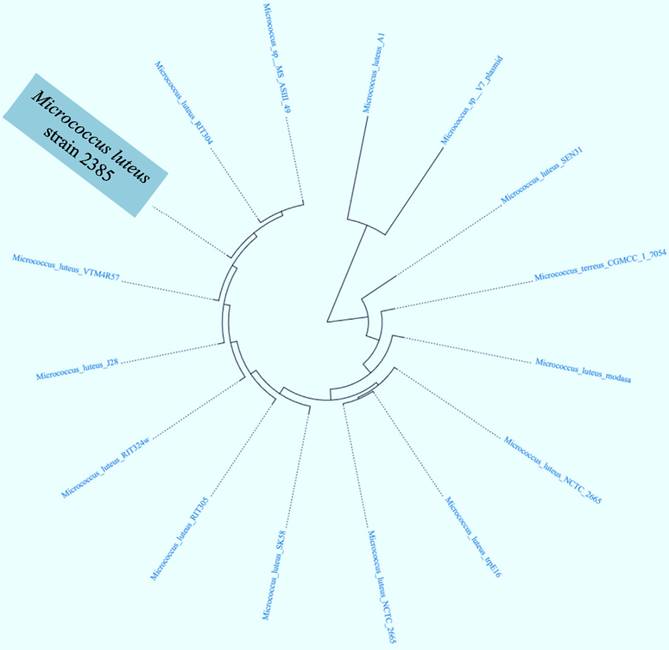
Further genome-wide analysis of strain 2385 revealed approximately 79.76% protein coding genes associating with function prediction, 33.99% genes were found connected to KEGG pathways and 69.89% genes associated with clusters of orthologous groups of proteins (COGs). We then selected the whole genome sequences of 15 other Micrococcus strains that are available in IMG/er to run a comparative analysis of strain 2385 relative to other sequenced Micrococcii, which revealed clustering with M. luteus strain VTM4R57, from rumen microbiome and M. luteus strain RIT304, which appears to be a plant endophyte, respectively (Fig. 2). Genome-wide comparison of M. luteus strain 2385 with other taxonomic relatives, including strains VTM4R57, J28, RIT304 and MS-ASIII-49 are shown in table 1.
Hierarchical clustering analysis based phylotree of Micrococcus sp. strain 2385 along with a cohort of Micrococcus spp., for which whole genome sequences are available The tree was generated based on the presence of Pfam category amongst Micrococcus spp. for which whole genome sequences are available in the IMG/er database.
General features of the genomes of Micrococcus species that taxonomically affiliated with the pesticide degrading Micrococcus sp. strain 2385.
| Characteristics | Micrococcus luteus strain 2385 | Micrococcus luteus VTM4R57 | Micrococcus luteus J28 | Micrococcus luteus RIT304 | Micrococcus sp. MS-ASIII-49 |
|---|---|---|---|---|---|
| NCBI No | 75105 | 1270 | 935864 | 1270 | 1593237 |
| Size (bp) | 2459555 | 2562419 | 2536485 | 2506829 | 2440202 |
| GC content (%) | 1791121 (72.82) | 1866350 (72.84) | 1843730 (72.69) | 1831038 (73.04) | 1783331 (73.02) |
| Total no. of Genes | 2268 | 2391 | 2428 | 2305 | 2342 |
| Protein Coding Genes | 2204 | 2330 | 2371 | 2248 | 2288 |
| Proteins with Function Prediction | 1806 | 1868 | 1882 | 1854 | 1829 |
| DNA Coding Density (%) | 2225045 (90.47) | 2339869 (91.31) | 2275198 (89.70) | 2217843 (88.47) | 2220480 (91.00) |
| Proteins assigned to COGs (%) | 1585 (69.89) | 1622 (67.84) | 1594 (65.65) | 1547 (67.11) | 1547 (66.05) |
| DNA Scaffolds | 175 | 37 | 83 | 183 | 183 |
| Total RNA Genes | 64 | 61 | 57 | 57 | 54 |
| tRNA Genes | 49 | 48 | 49 | 53 | 43 |
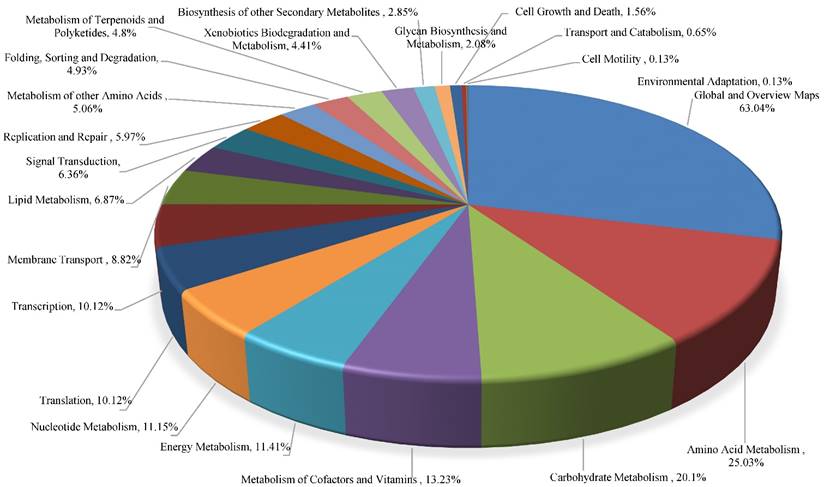
Furthermore, we performed a hierarchical clustering analysis based on the presence of categories of genes and genomes from KEGG (Kyoto Encyclopedia of Genes and Genomes), which revealed a plethora of functional traits identified in strain 2385. Of major note are a total of 88 genes identified for energy metabolism, (5.17% of its total genome) along with 34 genes for xenobiotics biodegradation and metabolism (2% of its total genome). Other examples of biodegradative genes found in the isolated strain included those involved in the pathways for the degradation of caprolactam, toluene, PAHs, and chloroaromatic compounds such as, chloroalkane, chloroalkene, and chlorobenzene, respectively.
Additionally, a total of 1585 protein coding genes associated with COGs in strain 2385 were found with further classification into 24 categories. The following were most abundant COG subsystems in strain 2385: amino acid transport and metabolism (n = 180); translation, ribosomal structure and biogenesis (n = 174); general function prediction only (n = 149); coenzyme transport and metabolism as well as inorganic ion transport and metabolism were found at the same level (n = 116). The strain also contains 10 biosynthetic gene clusters containing 142 genes; a total of 6.26% of the total genome size of strain 2385. These analyses suggest Micrococcus sp. strain 2385 to harbor various genome-enabled metabolic and catabolic processes.
Moreover, bacterial genomes are known to consist of a core set of genes encoding for essential metabolic functions, along with a plethora of genes that are acquired from the bacterium's native environment by horizontal gene transfer (HGT) mechanisms, providing the host with evolutionary adaptive traits and genome plasticity. Many such HGT-acquired genes occur as orthologous blocks commonly known as genomic islands (GEIs) 11. GEIs have been more commonly known to render virulence or antibiotic resistance to the host bacteria, but more recently, whole genome sequencing studies have also revealed other adaptive functional traits encoded by GEIs that can be further classified into the following 4 categories - pathogenicity islands (PAIs), that code for virulence genes; metabolic islands (MIs), genes for biosynthesis of secondary metabolites; resistance islands (RIs), genes that code for resistance- typically against antibiotics; and symbiotic islands (SIs), facilitating symbiotic associations of the host with other micro- and macroorganisms, respectively.
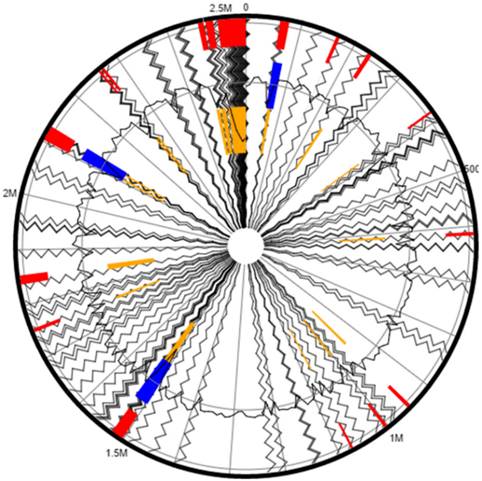
We used Island Viewer to identify genomic islands (GEIs) (http://www.pathogenomics.sfu.ca/islandviewer/) 12 in strain 2385. Island Viewer predicts GEIs integrating two widely used sequence composition based GEI prediction methods- SIGI-HMM and IslandPath-DIMOB along with a comparative GI prediction method- IslandPick. Interestingly, a total of 43 genomic islands were identified from the genome of Micrococcus sp. strain 2385 when aligned against the complete genome sequence of Micrococcus luteus NCTC 2665 as the reference strain (Fig. 4). When GEI-encoded gene homologues in strain 2385 were further analyzed by BLAST, several of them closely affiliated with genes previously shown active in catabolic processes, providing a strong evidence that the GEIs were horizontally acquired by strain 2385 from other biodegradative bacteria to facilitate survival in its native environment.
Functional traits found connected with KEGG pathways identified from the whole genome sequence of Micrococcus sp. strain 2385. The figure was prepared by analysis of the whole genome using IMG/er and plotted using Microsoft Excel.
Putative genomic islands (GEIs) predicted within the genome of Micrococcus sp. strain 2385 when aligned against the complete genome sequence of Micrococcus luteus NCTC 2665 as the reference. The outer black circle represents the scale line in Mbps and the black zig-zag line plot delineates each of the 175 contigs identified from strain 2385. GEIs obtained from each of the following methods are shown in color: SIGI-HMM (orange), IslandPath-DIMOB (blue), and integrated detection (red), respectively.
Nucleotide Sequence Accession Number. The draft genome sequence of Micrococcus sp. strain 2385 obtained in this study is available in NCBI Genome under the accession number of PRJNA265206 (http://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA265206); BioSample ID: SAMN03151542; WGS ID: JTKE00000000.
Acknowledgements
This work was jointly supported by the following grants: USAID-Egypt grant and the US Department of Defense (DoD) grants- W911NF-10-1-0146 and W911NF-10-R-0006. We also acknowledge funding provided by the Savannah River Nuclear Solutions under subcontract #0000217620 and the support provided by Dr. Stefan Green, University of Illinois at Chicago.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pimentel D. Environmental and Economic Costs of the Application of Pesticides Primarily in the United States. Integrated Pest Management: Innovation-Development Process. Volume 1. 2009 Editors: Dr. Rajinder Peshin, Ashok K. Dhawan
2. Alachlor in Drinking-water. Background document for development of WHO Guidelines for Drinking-water Quality. World Health Organization (WHO). 2003
3. Ewida YI. Biodegradation of Alachlor and Endosulfan Using Environmental Bacterial Strains. World Applied Sciences Journal. 2014;32(4):540
4. Khan JA, Singh S. Evaluation of oil degradation potential of Micrococcus varians. International Journal of Applied Biology and Pharmaceutical Technology. 2011;2:75-80
5. Nwankwegu AS, Onwosi CO, Orji MU, Anaukwu CG, Okafor UC, Azi F, Martins PE. Reclamation of DPK hydrocarbon polluted agricultural soil using a selected bulking agent. J Environ Manage. 2016;172:136-42
6. Zhang Y, Wang F, Zhu X, Zeng J, Zhao Q, Jiang X. Extracellular polymeric substances govern the development of biofilm and mass transfer of polycyclic aromatic hydrocarbons for improved biodegradation. Bioresour Technol. 2015;193:274-80
7. Chauhan A, Green S, Pathak A, Thomas J, Venkatramanan R. Whole-genome sequences of five oyster-associated bacteria show potential for crude oil hydrocarbon degradation. Genome Announc. 2013;3:1 (5)
8. Markowitz V M, Mavromatis M, Ivanova N N, Chen I-M A, Chu K, Kyrpides N C. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271-2278
9. Aziz R, Bartels D, Best A, DeJongh M, Disz T, Edwards R, Formsma K, Gerdes S, Glass E, Kubal M, Meyer F, Olsen G, Olson R, Osterman A, Overbeek R, McNeil L, Paarmann D, Paczian T, Parrello B, Pusch G, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST server: Rapid Annotations using Subsystems Technology. BMC Genomics. 2008;9:75
10. Grant JR, Arantes AS, Stothard P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics. 2012;13:202
11. Juhas M, van der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev. 2009;33(2):376-93
12. Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R, Nizam F, Pereira SK, Waglechner N, McArthur AG, Langille MG, Brinkman FS. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015;43(W1):W104-8
Author contact
![]() Corresponding author: Environmental Biotechnology and Genomics Laboratory, School of the Environment, 1515 S. MLK Blvd., Suite 305B FSHSRC, Florida A&M University, Tallahassee, FL-32307, USA; Phone: 850-412-5119, Fax: 850-561-2248, email: ashvini.chauhanedu.
Corresponding author: Environmental Biotechnology and Genomics Laboratory, School of the Environment, 1515 S. MLK Blvd., Suite 305B FSHSRC, Florida A&M University, Tallahassee, FL-32307, USA; Phone: 850-412-5119, Fax: 850-561-2248, email: ashvini.chauhanedu.