ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2018; 6:74-87. doi:10.7150/jgen.25648 This volume Cite
Research Paper
Genome Wide Analysis Reveals the Extrinsic Cellulolytic and Biohydrogen Generating Abilities of Neocallimastigomycota Fungi
Ayyappa Kumar Sista Kameshwar, Wensheng Qin ![]()
Department of Biology, Lakehead University, 955 Oliver Road, Thunder Bay, Ontario, P7B 5E1, Canada.
Received 2018-2-21; Accepted 2018-4-25; Published 2018-6-10
Abstract

Ruminating animals, especially cattle lack the carbohydrate active enzyme encoding genes which are required for the degradation of the glycosidic linkages of plant cell wall carbohydrates (such as cellulose, hemicellulose, lignin and pectin). Thus, ruminating animals are completely dependent on the microorganisms (anaerobic bacteria and fungi, methanogenic archaea and protozoa) residing in their rumen (hindgut). In this study, we have retrieved and analyzed the complete genome wide annotations of the Neocallimastigomycota division fungi such as Anaeromyces robustus, Neocallismatix californiae, Orpinomyces sp, Piromyces finnis, Piromyces sp E2. We have retrieved the InterPro, CAZy, KOG, KEGG, SM Clusters and MEROPS genome level data of these anaerobic fungi from JGI-MycoCosm database. Results obtained in our study reveals that, the genomes of anaerobic fungi completely lack genes encoding for lignin degrading auxiliary activity enzymes. Contrastingly, these fungi outnumbered other fungi by having highest number of CAZyme encoding genes. The genes encoding for dockerins and carbohydrate binding modules exaggerated other CAZymes which are involved in the structure and functioning of cellulosomes. Presence of cellulosomes and higher number of carbohydrate transport and metabolism genes also endorses the plant cell wall carbohydrate degrading abilities of these fungi. We also reported the tentative total cellulolytic, hemicellulolytic and pectinolytic abilities. And we have explicitly reported the genes, enzymes and the mechanisms involved in structure and functioning of the cellulosomes and hydrogenosomes. Our present work reveals the genomic machinery underlying the extrinsic plant cell wall degrading abilities of the anaerobic fungi. Results obtained in our study can be significantly applied in improving the gut health of cattle and especially in the fields of biofuel, biorefining and bioremediation-based industries.
Keywords: Ruminating animals (cattle), Neocallimastigomycota (Anaerobic fungi), Cellulose, Plant biomass, Cellulosomes, Hydrogenosomes
Introduction
Increasing global temperatures, over dependence and depletion of fossil fuels to meet the increasing fuel needs, have forced the mankind to produce sustainable renewable energy systems. Plant biomass which contains lignocellulose polymers constitutes the most abundant component on the earth's surface. Separation and production of renewable energy from the lignocellulosic biomass is a complex procedure and requires application of different chemical, physical and mechanical methods. However, biological way of biofuel production from lignocellulosic biomass is the most preferred and researched due to its ecofriendly and cost-effective nature. Naturally, enormous amounts of cellulose are digested by the herbivorous ruminating animals (having plant biomass containing diets e.g. cows, sheep, buffalo, sheep, deer, goats etc.) and this process of digestion is solely supported by microorganisms residing in its rumen. Gut of the ruminating animals can be divided into four chambers they are rumen, reticulum, omasum, abomasum (or stomach) (Figure S1). The process of food digestion starts from the rumen, saliva mixed food is mechanically broken down to smaller pieces which is passed into the reticulum where it separates the food into digestible and non-digestible forms into cuds (partly degraded food). The partially digested cuds are regurgitated, which is further rechewed and swallowed by the ruminating animals. Thus, from the partially digested food, omasum absorbs water, nutrients, vitamins, fatty acids etc. Major part of the cellulose fermentation happens in the rumen due to the presence of cellulolytic microorganisms. Studies have reported that anaerobic bacteria such as Ruminococcus genus (e.g. Ruminococcus albus, Ruminococcus flavefaciens) Streptococcus, Escherichia, Megasphaera, Fibrobacter are significantly involved in cellulose degradation [1-5].
In the year 1970, Colin Orpin has identified the anaerobic fungi based on its zoospores and chitin containing cell walls, which are now classified under the division of Neocallimastigomycota. This peculiar group of anaerobic fungi has raised considerable curiosity among mycologists worldwide. Two major reasons which brings Neocallimastigomycota fungi under the lime light are its distinctive physiology and potential applications in the fields of biomass conversion and animal nutrition [6]. Naturally these anaerobic fungi reside in the gastrointestinal tracts of herbivores and significantly aids in digestion of different plant biomass components. Anaerobic fungi were observed among all the foregut fermenters (where most of the digestion occurs before the gastric digestion) e.g. ruminants, pseudo-ruminants and foregut non-ruminants and also in most of the hindgut fermenters (where the most of the fermentation occurs after gastric digestion in the large intestine and caecum) [7]. Significantly, the anaerobic fungi residing in the gut of ruminating animals performs two major functions plant biomass digestion and forming a dedicated digestive chamber with neutral pH conditions [7]. Herbivorous animals lack the ability to digest plant biomass components as they cannot secrete cellulolytic/hemicellulolytic enzymes, in turn they depend on symbiotic gut microorganisms such as anaerobic bacteria and fungi, methanogenic archaea and protozoa [7]. Among the above-mentioned microorganisms, anaerobic fungi were found to play a crucial role in degradation of lignocellulosic components of plant biomass.
Studies conducted by Heath et al (1986), has revealed that reproduction in anaerobic fungi commences through asexual reproduction by releasing flagellated zoospores from sporangia[8]. Previous reports reveal that ingestion of food by the ruminating animals induces the anaerobic fungi to release its zoospores from the sporangia and it was also reported that within 30-60 minutes the density of zoospore peaks in the rumen [7, 9-12]. According to Orpin & Greenwood (1986), haem and other related porphyrin compounds released from the ingested plant materials trigger the sporangia of anaerobic fungi which induces the process of differentiation and maturation of zoospore in the rumen [13] (Figure S2). The flagellated zoospores of anaerobic fungi are motile and they colonize on the plant material based on the chemotactic responses from the surrounding sugars and phenolic compounds [14]. Flagellated zoospores transform to a cyst by shedding its flagella, once it attaches to the plant material. Formation and germination of cyst is involved by thickening of the cell wall and production of germ tube from the polar end and further development of cyst varies from monocentric to polycentric based on the organism. Endogenous cyst germination is observed in monocentric taxa as the nucleus is situated in the cyst forming a zoosporangium, by leaving rhizoids anucleate. Contrastingly, exogenous cyst germination is observed in polycentric taxa where the nucleus migrates to the rhizoids thus leading to the formation of multiple sporangia [7, 15]. The rhizomycelium (bulbous or filamentous) of the anaerobic fungi performs two major and important functions, it provides support to the growing sporangium (monocentric) or sporangia (polycentric) and performs the enzymatic digestion through penetrating into the plant material. Penetration of developing rhizoids into the rigid plant material, opens the internal plant tissues, making them susceptible to the enzymatic hydrolysis, which supports the developing sporangia with the nutrients for the development and maturation of the sporangia. Mature sporangia will produce zoospores ranging between 1 (less) to 80 (high), under suitable conditions (inducers), mature sporangium undergoes differentiation and further releases zoospores by dissolving the sporangial cell wall (Figure S2). Previous studies report that it is tough to free host animals from anaerobic fungi, as they exhibit efficient dispersal of anaerobic fungi among the host animals through forming aerotolerant cellular structures with extreme survival abilities (e.g. 2-4 chambered spores of Anaeromyces sp) [16, 17]. Studies have also confirmed that, anaerobic fungi can be cultured from the faecal material of host animals followed by air drying, freezing and from the long-settled cow dung [7, 18-20].
Studies conducted in the past have reported that axenic cultures of the anaerobic fungi produce a wide range of cellular end products of metabolism such as acetate, formate, lactate, hydrogen, carbondioxide and ethanol.[21, 22]. However, due to the process of hydrogen transfer associated with methanogens, thus presence of interspecies microbes alters the metabolic profile in the rumen [7, 23]. Recent genomic studies have successfully revealed the complete genomic sequences of anaerobic fungi Anaeromyces robustus, Neocallimastix californiae, Orpinomyces sp., Piromyces finnis and Piromyces sp E2 [24-27]. Genomic studies have concluded that due to the evolutionary loss of mitochondria these fungi have developed an efficient double membraned hydrogenosomes, which substitute mitochondria by generating ATP via substrate level phosphorylation and hydrogen [28-30]. Anaerobic fungi were also found to contain large multi-protein complexes called cellulosomes, which are involved in enzymatic hydrolysis of plant biomass. Understanding the genomic and proteomic abilities of these organisms encoding for the plant cell wall degrading enzymes will significantly help the biofuel and biorefining industries.
Industrially, anaerobic fungi exhibit various biotechnological applications especially applied as microbial probiotic supplement to improve the process of digestion and thus utilizing the low-quality forages. Several studies were already being conducted on improving the feed intake, efficiency, development-growth rate of animals and especially in milk production [31-34]. Previous findings suggest that, dietary supplementation of anaerobic fungal enzymes (e.g. glycoside hydrolases) in cattle resulted to be more effective than the viable cultures (e.g. swine and poultry). According to Azain et al., (2002), dietary supplementation of glycoside hydrolases (to support in depolymerization of plant biomass components) obtained from anaerobic fungi improved the growth and development of broiler chicken by 25% [7, 35]. Anaerobic fungi were of high interest in brewing, food, paper, textile and biofuel industries, due to its ability to secrete polysaccharide degrading enzymes. Studies were also being conducted for developing anaerobic fungi based and fermentative production of cellulosic ethanol and renewable fuels from the agricultural residues. Youssef et al., (2013) has reported the lignocellulose degradation study using Orpinomyces sp strain C1A by simultaneously producing cellulosic ethanol, Orpinomyces sp was found to degrade 61.3% of corn stover (dry weight) resulting in 0.045 to 0.096 mg of ethanol per mg of biomass [26]. In this study we have specifically analyzed the genome wide architecture of five completely sequenced genomes of anaerobic fungi classified under Neocallimastigomycota division. We have compared the genome wide lignocellulolytic abilities of these fungi specifically by comparing genome wide annotations such as CAZy, InterPro, MEROPS and SM (secondary metabolite) Clusters databases. Our study will reveal lignocellulolytic, detoxifying and degrading abilities of the anaerobic fungi.
Methods
Data retrieval
In our present study, we have selected and retrieved the genome level data of 5 anaerobic fungi classified under the Neocallimastigomycota division (Anaeromyces robustus v1.0 (Anasp1) [24], Neocallimastix californiae G1 v1.0 (Neosp1) [24], Orpinomyces sp. (Orpsp1_1) [26], Piromyces finnis v3.0 (Pirfi3) [24], Piromyces sp. E2 v1.0 (PirE2_1) [24]). The genome level data of all the above selected fungi were retrieved from the JGI-MycoCosm repository (https://genome.jgi.doe.gov/programs/fungi/index.jsf ).
Data Analysis
From JGI-MycoCosm database we have specifically selected and retrieved the genome wide annotations such as InterPro (a database for the protein families, domains and functional sites), CAZy (database for carbohydrate active enzymes), KOG (eukaryotic orthologous group) and SM Clusters (database for secondary metabolite gene clusters) of the above listed fungi. To understand and reveal the distribution of plant cell wall degrading enzymes and their evolutionary gene losses among the selected fungi. We have segregated and compared the total number of genome wide InterPro annotations of the selected fungi into protein domains occurring in a) multiple copies and b) single copies. Similarly, we have segregated and compared the total number of genes encoding for the glycoside hydrolases (GH), glycosyl transferases (GT), carbohydrate binding modules (CBM), auxiliary activity (AA), polysaccharide lyases (PL), dockerin (DOC) and expansin (EXPN) classes and total number of genome wide carbohydrate active enzymes among the selected fungi. We have also retrieved and compared the KOG (eukaryotic orthologous) groups a) cellular signaling and processing (CSP) b) information storage and processing (ISP) c) metabolism and d) poorly characterized. Finally, we have retrieved and compared the SM (secondary metabolite) clusters such as DMAT (Di Methyl Allyl Tryptophan Synthase), HYBRID (hybrid genes), NRPS (Non-Ribosomal Peptide Synthetases), Poly Ketide Synthases (PKS) and MEROPS (database for the peptidases) for comparing the distribution of proteases among the selected fungal genomes We have used Microsoft excel program for comparing and representing the genomes of the selected fungi. The analysis pipeline implemented in our present study is extensively represented in the Figure 1.
Workflow pipeline implemented for analyzing and comparing the genomes of Neocallimastigomycota division fungi
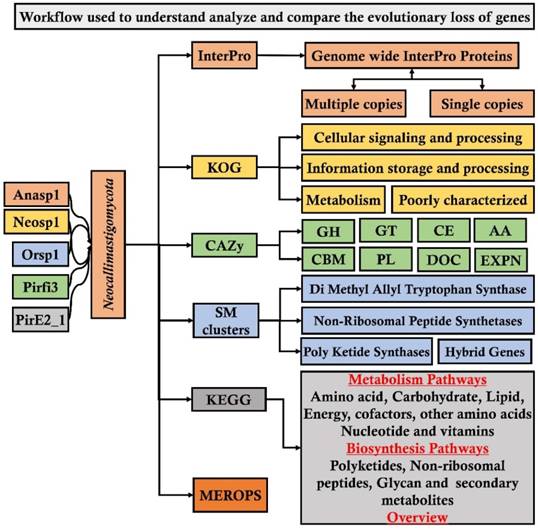
Results and Discussion
Genomic studies of Neocallimastigomycota fungi has revealed various interesting facts about its plant cell wall decaying abilities and about the evolutionary loss of several other genes involved in various metabolic processes. Neocallimastigomycota fungi were found to be closely related to the phylum Chytridiomycota. Though Neocallimastigomycota fungi share some of the key morphological characters as Chytridiomycota fungi, they exhibit some of the unique features such as their cellular physiology suitable for its anaerobic living and peculiar flagellar movement [7]. Later conducted genomic studies have endorsed the separation of Neocallimastigomycota into a separate clade basal to Chytridiomycota fungi [36-38]. Neocallimastigomycota fungi comprises of six different genera which can be separated based on its distinguishable morphological properties such as rhizoidal and bulbous morphologies of thallus and zoospore flagellation (mono vs polyflagellate) [7, 39, 40]. Currently, Neocallimastigomycota division comprises of nine genera Anaeromyces, Buwchfawromyces, Caecomyces, Cyllamyces, Neocallimastix, Oontomyces, Orpinomyces, Pecoramyces and Piromyces, where each genus possesses some unique and distinguishable morphological characteristics. Phylogenetic studies conducted in the past, based on the conserved DNA sequences such as 18S RNA and ITS (Inter transcribed spacer regions) sequences have reported the close relationship among Neocallimastix, Orpinomyces and Caecomyces, Cyllamyces genera respectively [7, 41, 42]. Recent whole genome sequencing studies of anaerobic fungi have reported the complete assembled and annotated genomes, with total number of assembled gene models raging between 10,992-Piromyces finnis, 12,832-Anaeromyces robustus, 14,648-Piromyces E2 18,936-Orpinomyces and 20,219-Neocallimastix californiae respectively [24, 26] (Figure 2).
(A) Hierarchical delineation of Neocallimastigomycota division and well-studied fungi with available annotated genomes, (B) phylogenetic relationship of the Neocallimastigomycota fungi based on the conserved ITS (Inter Transcribed Spacer regions) and other morphological characteristics, where the legend shows the phylogenetic relatedness among the selected fungi.
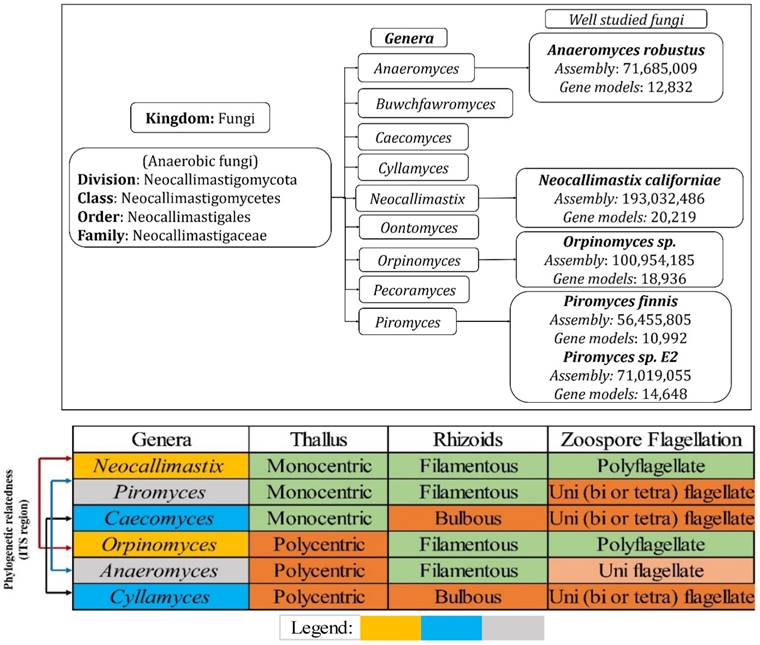
Genomes of Neocallimastigomycota fungi exhibited nearly about 3221 (Anasp1-Anaeromyces robustus v1.0 [24]), 3313 (Neosp1-Neocallimastix californiae G1 v1.0 [24]), 2598 (Orsp1-Orpinomyces sp. [26]), 3190 (Pirfi3-Piromyces finnis v3.0 [24]) and 2795 (Piromyces sp. E2 v1.0 [24]) unique InterPro annotated domains respectively (Figure S3A). Majority of these protein domains were found to occur in multiple copies in the genomes, making the genome wide distribution of InterPro protein domains to 30969 (Anasp1), 46794 (Neosp1), 35585 (Orsp1), 27050 (Pirf3) and 28577 (PirE2) respectively (Figure S3 B). Results obtained in our study reveals that genes encoding for ankyrin, WD-40 repeat, chitin-binding type-I, Src homology-3, cellulose binding region, protein kinases, leucine rich repeats, ABC transporters, dockerin (cellulose binding family 5), RNA recognition motif (RNP-1), calcium binding EF, Armadillo-type fold, serine/threonine protein kinase, zinc finger (RING, C2H2-type), tyrosine protein kinase, glycoside hydrolase, heat shock protein (DnaJ), tetratricopeptide region, AAA+ ATPase, spore coat protein and kinesin protein domain were observed in high numbers among the genomes of anaerobic fungi (Figure S3 B). To understand the genomic distribution of proteins we have analyzed the genome wide KOG annotations of the selected anaerobic fungi. We have retrieved the total number of gene models classified under cellular signaling and processing, information storage and processing, metabolism and poorly characterized KOG categories. The total KOG classified gene models where further analyzed to understand the proteins occurring in unique and multiple copies among the genomes of anaerobic fungi. Genes encoding for chitinases, leucine rich repeat, serine/threonine protein kinases, RNA-binding protein, lipid exporter, ankyrin have outnumbered all other protein encoding genes (Figure 3A). We have observed that Neosp1 genome harbors higher number of gene models compared to other anaerobic fungal genomes, followed by Orsp1genomes respectively (Figure 3A). The ascending order of the anaerobic fungi based on their total KOG classified gene models Pirfi3-8462 < Anasp1-9556 < PirE2-9810 < Orsp1-12559 < Neosp1-14,449 respectively (Figure 3B). Later, we have analyzed and classified total KOG proteins into a) unique protein coding genes and b) proteins with >2 gene copies (Figure 3C & 3D) respectively.
A) Genome wide distribution of KOG classified proteins under CSP (cellular signaling and processing), ISP (Information storage and processing), M (metabolism) and PC (poorly characterized) B) total number of KOG classified proteins C) unique genes D) proteins with multiple gene copies (>2 gene copies).

Results obtained from the classification of carbohydrate active enzymes (CAZymes) among Neocallimastigomycota (anaerobic) fungi, reveal that these fungi have suffered a severe evolutionary loss of genes encoding for lignin degrading enzymes. The total number of genes encoding for the auxiliary activity class enzymes such as lignin oxidizing enzymes (laccase, lignin peroxidase, manganese peroxidase, versatile peroxidase and cellobiose dehydrogenase) and lignin degrading auxiliary enzymes (aryl alcohol oxidase, vanillyl alcohol oxidases, glyoxal oxidases, pyranose oxidases, galactose oxidases, glucose oxidases and benzoquinone reductases) were completely reduced to zero. Importantly, genes encoding for GMC (glucose methanol choline) oxidoreductases, lytic polysaccharide monoxygenases (LPMO), cellobiose dehydrogenase (CDH) and iron reductase enzymes were completely reduced among the selected Neocallimastigomycota division fungi. Genes encoding for glycoside hydrolases, glycosyl transferases were found to range between 262-Anasp1(low) and 548-Neosp1 (high),104-Pirfi3(low) and 196-Neosp1 (high) respectively. While genes encoding for carbohydrate esterases, expansin and polysaccharide lyases ranges between 92-Pirfi3 (low) and 213-Neosp1(high), 9-Pirfi3 (low) and 29-Neosp1(high), 11-Anasp1(low) and 82-Neosp1(high) respectively (Figure 4A).
Genome wide distributions of A) CAZymes, B) cellulolytic, hemicellulolytic and pectinolytic activities (GH- Glycoside hydrolases, CE- Carbohydrate esterases and PL-polysaccharide lyases) where Anasp1 (Anaeromyces robustus), Neosp1 (Neocallimastix californiae), Orpsp1 (Orpinomyces sp), PirE2 (Piromyces sp. E2) and Pirfi3 (Piromyces finnis) respectively.
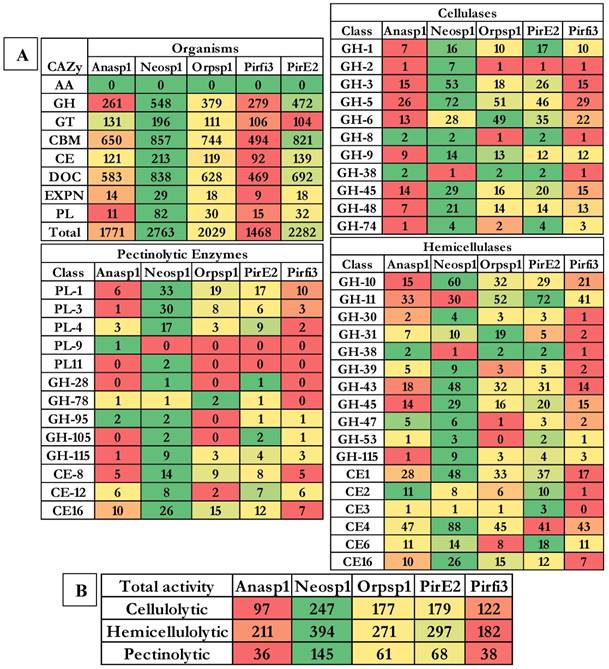
Contrastingly, the genomes of Neocallimastigomycota division fungi harbors higher number of genes encoding for carbohydrate binding domains (CBM) and dockerin proteins, which are found to be involved in the formation of cellulosomes (a complex structure involved in degradation of plant cell wall polysaccharides especially cellulose). The number of gene models encoding for CBM and dockerin proteins ranges between 494-Pirfi3 (low) and 857-Neosp1(high), 469-Pirfi3 (low) and 838-Neosp1(high) respectively. Totally, the number of CAZymes encoding genes ranges between Neosp1-2763 (high) and Pirfi3-1468 (low) respectively (Figure 4A). Studies have reported that anaerobic fungi residing in the animal guts are majorly involved in digestion of plant biomass components (cellulose, hemicellulose, pectin and lignin). Previous studies have classified the CAZyme classes coding for cellulases, hemicellulases, ligninases and pectinolytic enzymes [43, 44]. The glycoside hydrolase enzyme classes GH-1, GH-2, GH-3, GH-5, GH-6, GH-8, GH-9, GH-38, GH-45, GH-48 and GH-74 code for cellulases respectively. Based on the total number of cellulase encoding genes Anasp1 exhibits lower and Neosp1 exhibits higher cellulolytic activity comparatively. Glycoside hydrolase classes GH-10, GH-11, GH-30, GH-31, GH-38, GH-39, GH-43, GH-45, GH-47, GH-53, GH-115, and carbohydrate esterase class enzymes CE-1, CE-2, CE-3, CE-4, CE-6 and CE-16 codes for hemicellulases respectively. Based on the total number of hemicellulase encoding genes Pirfi3 exhibits lower and Neosp1 exhibits higher hemicellulolytic activity. Similarly, polysaccharide lyase class enzymes PL-1, PL-3, PL-4, PL-9, PL-11 glycoside hydrolase class enzymes GH-28, GH-78, GH-95, GH-105, GH-115 and carbohydrate esterase class enzymes CE-8, CE-12 and CE-16 are involved in degradation of pectin respectively. We have theoretically predicted the total cellulolytic, hemicellulolytic and pectinolytic enzyme activities by calculating the total number of cellulases, hemicellulases and pectinolytic enzymes. Anasp1 and Pirfi3 exhibits lower total cellulolytic, hemicellulolytic and pectinolytic activities. Neosp1 exhibits higher total cellulolytic, hemicellulolytic and pectinolytic activities, whereas Orsp1 and PirE2_1 exhibits total activities similarly (Figure 4B) (Figure S8 and S9).
The cluster of orthologous groups (COG) is a prokaryotic database used for the identification of ortholog and paralog proteins. Similarly, KOG the eukaryotic version for cluster of orthologous groups detects the ortholog and paralog proteins in the given eukaryotic genome. The total number of genes classified under each KOG category of the selected fungi were reported elaborately in supplementary information (Figure S5). The descending order of fungi based on their total number of KOG classified gene models are Neosp1>Orsp1>PirE2>Anasp1>Pirfi3 respectively (Figure 5A) The SM cluster database identifies the secondary metabolites of fungal genomes into three major classes as a) non-ribosomal peptide synthetases (NRPS) b) polyketide synthetases (PKS) and c) terpene synthetases (TS) respectively [45]. These key enzymes are involved in the production of the important fungal secondary metabolites a) non-ribosomal peptides and amino acid-derived compounds, (b) polyketides and fatty acid-derived compounds and (c) terpenes [45]. Using a set of modules (single ATC module or multimodule model with ATC repeating units) NRPS catalyzes the biosynthesis of small peptides (a ribosome independent mechanism) [46]. According to Finking and Marahiel (2004), three core domains (adenylation (A), thiolation (T) and a condensation domain (C)) containing module catalyzes the peptide bond formation on the megasynthase complex [47]. Polyketide synthetase (PKS) fungal modules containing three domains a) ketoacyl synthase (KS), b) acyl transferase and c) phosphopantetheine site [48] (Figure 5C).
Genome wide distributions of A) KOG (Eukaryotic orthologous groups), B) SM (secondary metabolite) clusters C) distribution of proteins among KEGG classified pathway groups and D) clan-based classification of proteolytic enzymes
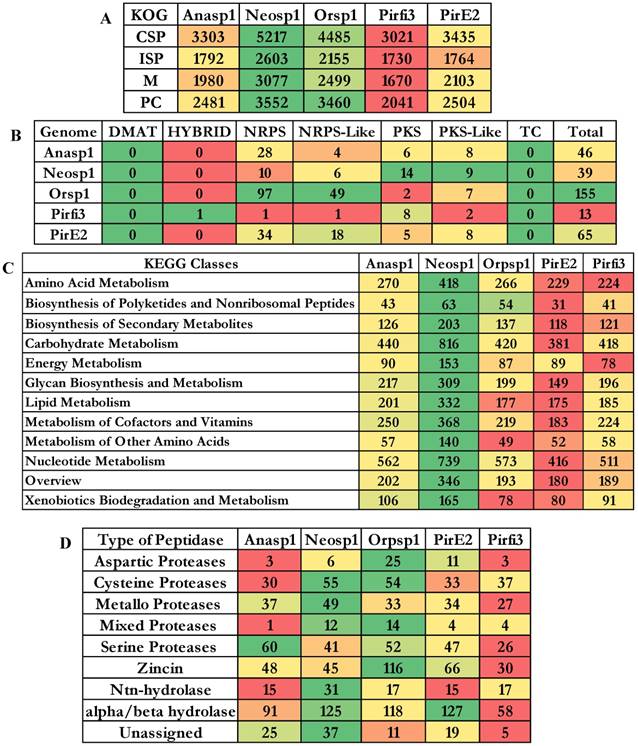
To broadly understand the genome wide distribution of genes involved in various cellular mechanisms, we have retrieved and compared the KEGG (Kyoto Encyclopedia of Genes and Genomes) annotations of the selected fungi. We have totally selected genes classified under 12 pathway classes (Figure 5B), out of these higher number of genes were classified under the carbohydrate and nucleotide metabolism pathways among all the selected anaerobic fungi. The descending order of the fungi based on the number of gene models classified among the 12 pathway classes were Neosp1>Orsp1>Anasp1>Pirfi3>PirE2_1 respectively (Figure 5B and Figure S7). The proteases (proteolytic enzymes or peptidases) play a crucial role in various molecular and biological processes. We have also analyzed the genome wide occurrence of proteolytic enzymes among the selected anaerobic fungi. MEROPS is public database for the proteolytic enzymes and their corresponding substrates and inhibitors respectively [49]. In MEROPS the proteolytic enzymes are classified based on its similitude of the protein structure at the tertiary and primary levels by specifically comparing the active and reactive sites of the proteases. The proteases are classified into families and clans respectively. Results obtained in our analysis shows that aspartic, mixed and zincin class proteases were high in Orsp1, cysteine, mixed, metallo, Ntn-hydrolase, alpha-beta hydrolase class proteases were high in Neosp1, serine proteases are high in Anasp1 and alpha/beta hydrolase protases were high in PirE2 genomes respectively (Figure 5D and Figure S6).
Our analysis explicitly reports that the selected anaerobic fungi harbors large number of genes encoding for the carbohydrate transport and metabolism. Thus, we have specifically focused on the total number of genes involved in carbohydrate transport and metabolism (Figure S4). Interestingly, we have observed that genomes of the selected anaerobic fungi harbor higher number of chitinase encoding gene models ranging between 293 (Pirfi3) to 639 (Neosp1). Previous studies have already reported that amino acid tryptophan was found to conserved in the chitin binding domain in bacteria, these domains were also found to be involved during binding of cellulase with cellulose [50]. For our present analysis we have only considered only the enzymes with multiple gene copies (<2). We have observed that 20 protein encoding gene models involved in carbohydrate transport and metabolism were found to be occur in multiple copies in the genomes of the selected anaerobic fungi genomes (Figure S3). Genes encoding for β-1,6-N-acetylglucosaminyltransferase, gluconate transport-inducing protein, β-glucosidase, Golgi mannosyltransferase, maltase glucoamylase (GH-31) were found to occur more than five copies in the genomes of anaerobic fungi respectively (Figure S2). Occurrence of higher number of gene models encoding for the carbohydrate transport metabolism proteins the ascending order based on the genomes were 492 (Pirfi3) > 578 (Anasp1) > 676 (Orsp1) > 693 (PirE2_1) > 1037 (Neosp1) respectively. These genomic evidences suggest that anaerobic fungi classified under Neocallimastigomycota has developed sophisticated organelles such as cellulosomes and hydrogenosomes for the degradation of plant cell wall components [24, 51].
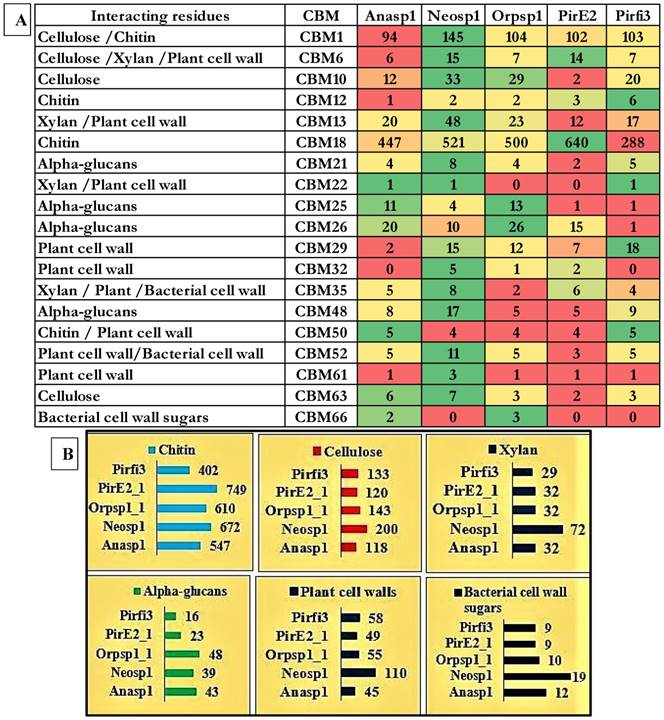
To understand the carbohydrate breakdown and metabolism by the Neocallimastigomycota division fungi, we have analyzed and compared the genomes of the selected anaerobic fungi to reveal the carbohydrate binding modules (CBM) and the corresponding carbohydrate interacting residues (Figure 6A). Our results report that genes encoding for carbohydrate binding modules (CBM18) interacting with chitin residues were found to be high in PirE2_1-640 and low in Pirfi3-288 respectively. We have analyzed and compared the total number of genes encoding for the carbohydrate binding module classes interacting with cellulose residues such as CBM-1, CBM-6 CBM-10, CBM-63. Similarly, we have analyzed and compared the CBM's interacting with xylan (CBM6, CBM13, CBM22 and CBM35) plant cell walls (CBM6, CBM13, CBM22, CBM29, CBM32, CBM35, CBM50, CBM52 and CBM61), alpha-glucans (CBM21, CBM25, CBM26 and CBM48) and chitin (CBM1, CBM12, CBM18 and CBM50) residues respectively. We have tentatively calculated the total number of genes encoding for CBM interacting with cellulose, xylan, chitin, alpha-glucans, plant cell wall and bacterial cell wall sugars. These results suggest that Neosp1 harbors higher number of cellulose, xylan, plant cell wall sugar binding CBM, whereas Orsp1 contains higher number of alpha glucan binding CBM and PirE2 contains higher number of chitin binding CBMs respectively (Figure 6B). Thus, anaerobic fungal resides higher number of genes encoding for the carbohydrate binding modules, dockerin proteins involved in maintenance of the structure and functioning of cellulosomes.
A) Carbohydrate binding modules (CBM) and the corresponding carbohydrate interacting residues, B) total tentative number of genes encoding for cellulose, xylan, alpha-glucans, plant cell walls and chitin binding CBM.
Recent genomic studies of the anaerobic fungi conducted by Haitjema, C.H et al (2017), have clearly proved the occurrence of 1600 dockerin domain proteins (DDP). About 20% of these dockerin domain proteins can be classified under CotH spore coat protein, these proteins were expected to be involved in binding with the plant cell wall components however, the exact function of it is not known till today [24]. Studies have also reported that majority of the lignocellulolytic enzymes contain non-catalytic dockerin domains (NCDDs) which facilitate the assembly of multiprotein cellulosome complexes and further required for the binding of carbohydrates and degradation of plant biomass [24]. It was reported that anaerobic fungal cellulosomes exhibits 13% higher GH activity due to the presence of glycoside hydrolase classes GH-3, GH-6 and GH-45 compared to bacterial cellulosomes. Especially the supplementary GH-3 class enzyme (Beta glucosidase) activity empowers fungal cellulosomes in converting cellulose to single fermentable sugars (monosaccharides) when compared to low molecular weight oligosaccharides generating bacterial cellulosomes (eg: Clostridium sp) [24]. Thus, the complete genome sequencing studies of Anaeromyces robustus (Anasp1), Neocallimastix californiae (Neosp1) and Piromyces finnis (Pirf3) by Haitjema, C.H et al (2017) and Orpinomyces sp Strain C1A (Orsp1) by Youssef, N. H et al (2013), have clearly reported that genomic sequences of fungal dockerin and scaffoldin proteins are not like that of bacterial cellulosome components. Which explains that the gut residing anaerobic fungi have exclusively evolved (bacterial independent) in developing the cellulosome based degradation of plant cell wall components [24, 26].
The anaerobic fungi of Neocallimastigomycota division were also found to possess an efficient enzyme system involved in structural and functional maintenance of hydrogenosomes respectively. Hydrogenosomes were observed in distantly related unicellular microorganisms, it is possible that hydrogenosomes have progressively evolved under various evolutionary conditions from the already existing mitochondria [27]. According to Boxma et al (2004), anaerobic fungal hydrogenosomes are significantly evolved from its counterparts (other anaerobic microorganisms) in various aspects including the underlying mechanisms required to catabolize pyruvate and final products generated from fermentation [52]. In general, anaerobic microorganisms contain hydrogenosomes (intracellular membrane bound organelles) in replacement of mitochondria of aerobic microorganisms [27]. In anaerobic microorganisms hydrogenosomes are also involved in conversion of fermentable monosaccharides released from cellulose breakdown to hydrogen gas through glycolysis. Hydrogenosomes in general metabolize malate, pyruvate to formate, acetate, hydrogen, carbondioxide and ATP (energy) [53] (Figure 7A). Giezen et al (1997) have reported that, hydrogenosomes of anaerobic fungi have originated from the mitochondria due to presence of targeting signal on the malic enzyme characterized from the hydrogenosome of Neocallimastix frontalis [27, 54].
A) Schematic tentative representation of genes involved in carbohydrate metabolism and hydrogenosomes reaction mechanisms employed by anaerobic fungi, B) Heatmap showing the tentative list of genes involved in hydrogenosomes and its related mechanisms
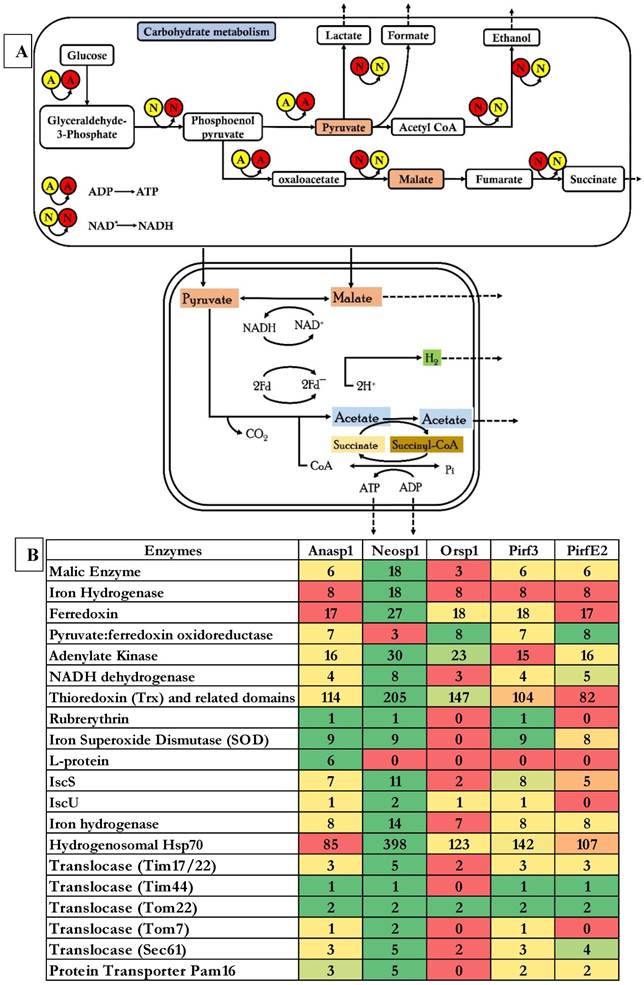
Studies have also reported that hydrogenosomes also provides anaerobic fungi with various other advantages a) it was reported that hydrogenosomes of Neocallimastix sp. L2 supports in survival of this fungi by scavenging the oxygen [55]. It was also reported that microorganisms reduce the hydrogen produced from the hydrogenosomes to more feasible methane gas. Thus, hydrogen transfer system between the species helps in maintaining the thermodynamic equilibrium of the gut flora and also in the cellulose breakdown by anaerobic fungi. While the methanogen co-culturing experiment conducted by Marvin-Sikkema et al (1990), have resulted in enhanced anaerobic fungi's cellulolytic activity, by resulting in metabolic shift of decreased hydrogen, lactate, succinate and ethanol production followed by increased methane and acetate productions [56]. Earlier studies have clearly classified the metabolic mechanisms involved in functioning of the hydrogenosomes majorly into a) ATP production b) Fe-S cluster biosynthesis and c) oxygen stress response [53, 57-60]. We have observed that the selected genomes exhibited the InterPro domains encoding for malic enzyme, pyruvate ferredoxin oxidoreductase, ferredoxin 2Fe-2S, succinate thiokinase, adenylate kinase, hydrogenase, succinate coA transferase respectively. Similarly, protein domains encoding for Fe-S cluster mechanisms such as iron-sulfur cluster (ISC) proteins IscU, IscA, IscS, ferredoxin, chaperons (heat shock proteins) Hsp70, Hsp60, Hsp10. Protein domains encoding for iron dependent superoxide dismutase, thioredoxin, NADH dehydrogenase, rubrerythrin, serine hydroxymethyl transferase and peptidase such as hydrogenosome processing peptidases, hydrogenosome membrane proteins were also observed. Apart from these protein domains there are certain number of membrane proteins called translocases which helps in movement of the proteins across the membranes such as translocases of outer (TOM), translocases of inner membranes, motor complex proteins and putative members of translocation machinery respectively [61]. We have compared the genomes of the five selected anaerobic fungi to list the enzymes involved in the metabolic mechanisms of the hydrogenosomes structure and functioning (Figure 7B) (Figure S10).
Conclusion
Genetically cattle lack the ability to encode for the lignocellulolytic enzymes, thus they are solely dependent on their rumen microbiota for the degradation of plant cell wall components. Improving the feeding efficiency and digestibility of the low-quality plant biomass components by manipulating the rumen microbiota and rumen fermentation is gaining its prominence in the recent times. Studies were being conducted to increase the number of lignocellulolytic microorganisms in rumen and their overall catalytic efficiency respectively [62]. Several microorganisms including bacteria (Ruminococcus genus, Megasphaera, Fibrobacter, Streptococcus, Escherichia), archaea (methanogens), fungi (Chytridiomycetes and Neocallimastigomycetes) were observed among the rumen. However, earlier studies have predicted that about 70% of the rumen microbiota is still unknown till today [63]. Anaerobic fungi potentially secrete higher number of lignocellulolytic enzymes such as cellulase (microcrystalline cellulose), xylanase, pectinase and proteases respectively. Anaerobic fungi degrade the plant biomass by breaking the fibrous plant components through penetrating its rhizoids and facilitate the access for the rumen microorganisms to the secondary cell wall components, thus playing a crucial role in degradation of the poor quality plant cell wall components.[64-66].
In this study we have analyzed and compared the genomic properties of the recently sequenced five anaerobic fungi. The genome level data and the corresponding annotations were retrieved from the JGI-MycoCosm database. Genome level annotations of the selected anaerobic fungi were compared among the Neocallimastigomycota division fungi. We have observed that several genes and protein domains occurred in multiple copies among the genomes of the selected anaerobic fungi. Genome of Neocallimastix californiae outnumbered other anaerobic fungi based on the total number of genes categorized under CAZy, SM Clusters, KOG and thus the KEGG pathways. Anaerobic fungi encode for all the genetic material necessary for the structure and functioning of hydrogenosomes, as these fungi lack mitochondria making them solely dependent on the hydrogenosomes. Similarly, these fungi also encode for higher number of genes involved in carbohydrate transport and metabolism with most of the genes (carbohydrate binding modules and dockerins) involved in formation of complex multienzyme organelles employed for efficient degradation of plant cell wall carbohydrates. Using the results obtained we have tentatively calculated the degradation potentials of these fungi, which suggests that Neosp1 possess highest cellulolytic, hemicellulolytic and pectinolytic abilities than other anaerobic fungi. We have also tentatively reported the mechanism involved during the carbohydrate metabolism by these fungal organelles respectively. In this study we have specifically emphasized on the genomic properties of the anaerobic fungi by analyzing the genome wide annotations of these fungi. Understanding the genomic complexities of anaerobic fungi will significantly enhance the gut health of the cattle (increases the total enzyme activity) and also supports the biofuel and biorefining industries in making eccentric enzyme mix with efficient catalytic properties.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by Natural Sciences and Engineering Research Council of Canada Funding (RGPIN-2017-05366) to Wensheng Qin and Ontario Trillium Scholarship (OTS) to Ayyappa Kumar Sista Kameshwar.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Leschine SB. Cellulose degradation in anaerobic environments. Annual Reviews in Microbiology. 1995;49:399-426
2. Brulc JM, Yeoman CJ, Wilson MK, Miller MEB, Jeraldo P, Jindou S. et al. Cellulosomics, a gene-centric approach to investigating the intraspecific diversity and adaptation of Ruminococcus flavefaciens within the rumen. PLoS One. 2011;6:e25329
3. Weimer PJ. Cellulose degradation by ruminal microorganisms. Critical Reviews in Biotechnology. 1992;12:189-223
4. Wallace RJ. Gut microbiology-broad genetic diversity, yet specific metabolic niches. Animal. 2008;2:661-8
5. Morrison M, Miron J. Adhesion to cellulose by Ruminococcus albus: a combination of cellulosomes and Pil-proteins? FEMS microbiology letters. 2000;185:109-15
6. Griffith GW, Baker S, Fliegerova K, Liggenstoffer A, van der Giezen M, Voigt K. et al. Anaerobic fungi: Neocallimastigomycota. IMA fungus. 2010;1:181-5
7. Gruninger RJ, Puniya AK, Callaghan TM, Edwards JE, Youssef N, Dagar SS. et al. Anaerobic fungi (phylum Neocallimastigomycota): advances in understanding their taxonomy, life cycle, ecology, role and biotechnological potential. FEMS microbiology ecology. 2014;90:1-17
8. Heath IB, Kaminskyj S, Bauchop T. Basal body loss during fungal zoospore encystment: evidence against centriole autonomy. Journal of cell science. 1986;83:135-40
9. Orpin C. Studies on the rumen flagellate Neocallimastix frontalis. Microbiology. 1975;91:249-62
10. Orpin C. Studies on the rumen flagellate Sphaeromonas communis. Microbiology. 1976;94:270-80
11. Orpin C. The occurrence of chitin in the cell walls of the rumen organisms Neocallimastix frontalis, Piromonas communis and Sphaeromonas communis. Microbiology. 1977;99:215-8
12. Orpin C. The rumen flagellate Piromonas communis: its life-history and invasion of plant material in the rumen. Microbiology. 1977;99:107-17
13. Orpin CG, Greenwood Y. The role of haems and related compounds in the nutrition and zoosporogenesis of the rumen chytridiomycete Neocallimastix frontalis H8. Microbiology. 1986;132:2179-85
14. Wubah D, Kim D. Isolation and characterisation of a free-living species of Piromyces from a pond. Abstracts of the Mycological Society of America (Inoculum). 1996
15. Trinci AP, Davies DR, Gull K, Lawrence MI, Nielsen BB, Rickers A. et al. Anaerobic fungi in herbivorous animals. Mycological Research. 1994;98:129-52
16. Becker ER. Methods of rendering the rumen and reticulum of ruminants free from their normal infusorian fauna. Proceedings of the National Academy of Sciences. 1929;15:435-8
17. Brookman JL, Ozkose E, Rogers S, Trinci AP, Theodorou MK. Identification of spores in the polycentric anaerobic gut fungi which enhance their ability to survive. FEMS microbiology ecology. 2000;31:261-7
18. Lowe SE, Theodorou MK, Trinci AP. Isolation of anaerobic fungi from saliva and faeces of sheep. Microbiology. 1987;133:1829-34
19. Milne A, Theodorou MK, Jordan MG, King-Spooner C, Trinci AP. Survival of anaerobic fungi in feces, in saliva, and in pure culture. Experimental Mycology. 1989;13:27-37
20. Davies DR, Theodorou MK, Lawrence MI, Trinci AP. Distribution of anaerobic fungi in the digestive tract of cattle and their survival in faeces. Microbiology. 1993;139:1395-400
21. Bauchop T, Mountfort DO. Cellulose fermentation by a rumen anaerobic fungus in both the absence and the presence of rumen methanogens. Applied and Environmental Microbiology. 1981;42:1103-10
22. Cheng YF, Edwards JE, Allison GG, Zhu W-Y, Theodorou MK. Diversity and activity of enriched ruminal cultures of anaerobic fungi and methanogens grown together on lignocellulose in consecutive batch culture. Bioresource technology. 2009;100:4821-8
23. Voncken F, Boxma B, Tjaden J, Akhmanova A, Huynen M, Verbeek F. et al. Multiple origins of hydrogenosomes: functional and phylogenetic evidence from the ADP/ATP carrier of the anaerobic chytrid Neocallimastix sp. Molecular microbiology. 2002;44:1441-54
24. Haitjema CH, Gilmore SP, Henske JK, Solomon KV, de Groot R, Kuo A. et al. A parts list for fungal cellulosomes revealed by comparative genomics. Nature microbiology. 2017;2:17087
25. Mondo SJ, Dannebaum RO, Kuo RC, Louie KB, Bewick AJ, LaButti K. et al. Widespread adenine N6-methylation of active genes in fungi. Nature Genetics. 2017:49
26. Youssef NH, Couger M, Struchtemeyer CG, Liggenstoffer AS, Prade RA, Najar FZ. et al. The genome of the anaerobic fungus Orpinomyces sp. strain C1A reveals the unique evolutionary history of a remarkable plant biomass degrader. Applied and environmental microbiology. 2013;79:4620-34
27. Haitjema CH, Solomon KV, Henske JK, Theodorou MK, O'Malley MA. Anaerobic gut fungi: advances in isolation, culture, and cellulolytic enzyme discovery for biofuel production. Biotechnology and bioengineering. 2014;111:1471-82
28. Mentel M, Martin W. Energy metabolism among eukaryotic anaerobes in light of Proterozoic ocean chemistry. Philosophical Transactions of the Royal Society of London B: Biological Sciences. 2008;363:2717-29
29. Müller M, Mentel M, van Hellemond JJ, Henze K, Woehle C, Gould SB. et al. Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiology and Molecular Biology Reviews. 2012;76:444-95
30. Tachezy J, Doležal P. Iron-sulfur proteins and iron-sulfur cluster assembly in organisms with hydrogenosomes and mitosomes. Origin of mitochondria and hydrogenosomes: Springer. 2007:105-33
31. Dey A, Sehgal JP, Puniya AK, Singh K. Influence of an anaerobic fungal culture (Orpinomyces sp.) administration on growth rate, ruminal fermentation and nutrient digestion in calves. ASIAN AUSTRALASIAN JOURNAL OF ANIMAL SCIENCES. 2004;17:820-4
32. Lee S, Ha J, Cheng K-J. Influence of an anaerobic fungal culture administration on in vivo ruminal fermentation and nutrient digestion. Animal Feed Science and Technology. 2000;88:201-17
33. Paul SS, Deb SM, Punia BS, Das KS, Singh G, Ashar MN. et al. Effect of feeding isolates of anaerobic fungus Neocallimastix sp. CF 17 on growth rate and fibre digestion in buffalo calves. Archives of animal nutrition. 2011;65:215-28
34. Gao AW, Wang HR, Yang JL, Shi CX. The effects of elimination of fungi on microbial population and fiber degradation in sheep rumen. Applied Mechanics and Materials: Trans Tech Publ. 2013:224-31
35. Azain M, Li X, Shah A, Davies T. Separation of the effects of xylanase and b-glucanase addition on performance of broiler chicks fed barley based diets. Int Poult Sci Forum. 2002:111
36. Fliegerova K, Hodrova B, Voigt K. Classical and molecular approaches as a powerful tool for the characterization of rumen polycentric fungi. Folia microbiologica. 2004;49:157-64
37. James TY, Kauff F, Schoch CL, Matheny PB, Hofstetter V, Cox CJ. et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature. 2006;443:818
38. James TY, Letcher PM, Longcore JE, Mozley-Standridge SE, Porter D, Powell MJ. et al. A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia. 2006;98:860-71
39. Ho Y, Barr D. Classification of anaerobic gut fungi from herbivores with emphasis on rumen fungi from Malaysia. Mycologia. 1995:655-77
40. Ozkose E. Morphology and molecular ecology of anaerobic fungi. University of Wales, Aberyswyth. 2001
41. Brookman J, Mennim G, Trinci A, Theodorou M, Tuckwell D. Identification and characterization of anaerobic gut fungi using molecular methodologies based on ribosomal ITS1 and 18S rRNA. Microbiology. 2000;146:393-403
42. Hausner G, Inglis G, Yanke L, Kawchuk L, McAllister T. Analysis of restriction fragment length polymorphisms in the ribosomal DNA of a selection of anaerobic chytrids. Canadian Journal of Botany. 2000;78:917-27
43. Sista Kameshwar AK, Qin W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology. 2017:1-13
44. Kameshwar AKS, Qin W. Metadata Analysis of Phanerochaete chrysosporium gene expression data identified common CAZymes encoding gene expression profiles involved in cellulose and hemicellulose degradation. International journal of biological sciences. 2017;13:85
45. Hoffmeister D, Keller NP. Natural products of filamentous fungi: enzymes, genes, and their regulation. Natural product reports. 2007;24:393-416
46. Bushley KE, Turgeon BG. Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC evolutionary biology. 2010;10:26
47. Finking R, Marahiel MA. Biosynthesis of nonribosomal peptides. Annu Rev Microbiol. 2004;58:453-88
48. Kroken S, Glass NL, Taylor JW, Yoder O, Turgeon BG. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proceedings of the National Academy of Sciences. 2003;100:15670-5
49. Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic acids research. 2013;42:D503-D9
50. Hamid R, Khan MA, Ahmad M, Ahmad MM, Abdin MZ, Musarrat J. et al. Chitinases: an update. Journal of pharmacy & bioallied sciences. 2013;5:21
51. van der Giezen M, Sjollema KA, Artz RR, Alkema W, Prins RA. Hydrogenosomes in the anaerobic fungus Neocallimastix frontalis have a double membrane but lack an associated organelle genome. FEBS letters. 1997;408:147-50
52. Boxma B, Voncken F, Jannink S, Van Alen T, Akhmanova A, Van Weelden SW. et al. The anaerobic chytridiomycete fungus Piromyces sp. E2 produces ethanol via pyruvate: formate lyase and an alcohol dehydrogenase E. Molecular microbiology. 2004;51:1389-99
53. Müller M. The hydrogenosome. Microbiology. 1993;139:2879-89
54. Van Der Giezen M, Rechinger KB, Svendsen I, Durand R, Hirt RP, Fevre M. et al. A mitochondrial-like targeting signal on the hydrogenosomal malic enzyme from the anaerobic fungus Neocallimastix frontalis: support for the hypothesis that hydrogenosomes are modified mitochondria. Molecular microbiology. 1997;23:11-21
55. Marvin-Sikkema FD, Gomes TMP, Grivet J-P, Gottschal JC, Prins RA. Characterization of hydrogenosomes and their role in glucose metabolism of Neocallimastix sp. L2. Archives of microbiology. 1993;160:388-96
56. Marvin-Sikkema F, Richardson A, Stewart C, Gottschal J, Prins R. Influence of hydrogen-consuming bacteria on cellulose degradation by anaerobic fungi. Applied and Environmental Microbiology. 1990;56:3793-7
57. Tachezy J, Sánchez LB, Müller M. Mitochondrial type iron-sulfur cluster assembly in the amitochondriate eukaryotes Trichomonas vaginalis and Giardia intestinalis, as indicated by the phylogeny of IscS. Molecular Biology and Evolution. 2001;18:1919-28
58. Coombs GH, Westrop GD, Suchan P, Puzova G, Hirt RP, Embley TM. et al. The amitochondriate eukaryote Trichomonas vaginalis contains a divergent thioredoxin-linked peroxiredoxin antioxidant system. Journal of Biological Chemistry. 2004;279:5249-56
59. Sutak R, Dolezal P, Fiumera HL, Hrdy I, Dancis A, Delgadillo-Correa M. et al. Mitochondrial-type assembly of FeS centers in the hydrogenosomes of the amitochondriate eukaryote Trichomonas vaginalis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10368-73
60. Pütz S, Gelius-Dietrich G, Piotrowski M, Henze K. Rubrerythrin and peroxiredoxin: two novel putative peroxidases in the hydrogenosomes of the microaerophilic protozoon Trichomonas vaginalis. Molecular and biochemical parasitology. 2005;142:212-23
61. Schneider RE, Brown MT, Shiflett AM, Dyall SD, Hayes RD, Xie Y. et al. The Trichomonas vaginalis hydrogenosome proteome is highly reduced relative to mitochondria, yet complex compared with mitosomes. International journal for parasitology. 2011;41:1421-34
62. Paul SS, Deb SM, Punia BS, Singh D, Kumar R. Fibrolytic potential of anaerobic fungi (Piromyces sp.) isolated from wild cattle and blue bulls in pure culture and effect of their addition on in vitro fermentation of wheat straw and methane emission by rumen fluid of buffaloes. Journal of the Science of Food and Agriculture. 2010;90:1218-26
63. Cho S-J, Cho K-M, Shin E-C, Lim W-J, Hong S-Y, Choi B-R. et al. 16S rDNA Analysis 9f Bacterial Diversity in Three Fractions of Cow Rumen. Journal of Microbiology and Biotechnology. 2006;16:92-101
64. Orpin CG. Anaerobic fungi: taxonomy, biology and distribution in nature. Anaerobic fungi: biology, ecology and function. 1994;12:1-45
65. Gordon G, Phillips MW. Degradation and utilization of cellulose and straw by three different anaerobic fungi from the ovine rumen. Applied and Environmental Microbiology. 1989;55:1703-10
66. Grenet E, Breton A, Barry P, Fonty G. Rumen anaerobic fungi and plant substrate colonization as affected by diet composition. Animal Feed Science and Technology. 1989;26:55-70
Author contact
![]() Corresponding author: Email: wqinca, Tel: 807-343-8467
Corresponding author: Email: wqinca, Tel: 807-343-8467