ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2018; 6:127-132. doi:10.7150/jgen.27864 This volume Cite
Short Research Paper
De novo male gonad transcriptome draft for the marine mussel Perumytilus purpuratus with a focus on its reproductive-related proteins
Carolina Briones1 ![]() , José J. Nuñez1, Montse Pérez2, Daniela Espinoza-Rojas3, Cristian Molina-Quiroz4, Ricardo Guiñez5
, José J. Nuñez1, Montse Pérez2, Daniela Espinoza-Rojas3, Cristian Molina-Quiroz4, Ricardo Guiñez5
1. Instituto de Ciencias Marinas y Limnológicas, Universidad Austral de Chile, Casilla 567, Valdivia, Chile
2. Instituto Español de Oceanografía, Centro Oceanográfico de Vigo, 36200 Vigo, España
3. Dirección de Calidad de Pregrado, Universidad de Talca, 2 Norte 685, Talca, Chile
4. Lircaytech, Irarrázabal 3782, Ñuñoa, Santiago de Chile
5. Instituto de Ciencias Naturales Alexander von Humboldt, Facultad de Ciencias del Mar y Recursos Biológicos, Universidad de Antofagasta, Casilla 170, Antofagasta, Chile
Received 2018-6-13; Accepted 2018-9-4; Published 2018-11-16
Abstract

Perumytilus purpuratus is a marine mussel considered a bioengineer species with a broad distribution in the Pacific and Atlantic coast of South America. Studies have shown two geographically and genetically differentiated subpopulations at molecular level and in sperm morphological traits. To open avenues for molecular research on P. purpuratus, a global de novo transcriptome from gonadal tissue of mature males was sequenced using the Illumina platform. From a total of 126.38 million reads, 37,765 transcripts were successfully annotated. BUSCO analysis determined a level of 89% completeness for the assembled transcriptome. The functional gene ontology (GO) annotation indicated that, in terms of abundance, the transcripts related with molecular function were the most represented, followed by those related with biological process and cellular components. Additionally, a subset of GO annotations generated using the “sperm” term resulted in a total of 1,294 sequences where the biological process category was the more represented, with transcripts strongly associated to sperm-processes required for fertilization, and with processes where the sperm-egg interaction could be implicated.
Our work will contribute to the evolutionary understanding of the molecular mechanisms related to tissue-specific functions. This work reports the first male gonad transcriptome for the mussel P. purpuratus, generating a useful transcriptomic resource for this species and other closely related mytilids.
Keywords: Gene Ontology, Mollusca, Bivalve, Sperm, Gamete-recognition proteins, BUSCO
Introduction
The marine mussel Perumytilus purpuratus (Mytilidae) is a dominant competitor and an ecosystem engineer forming extensive beds in the intertidal rocky zone [1]. This species inhabits throughout a broad latitudinal distribution comprising the Pacific Ocean, from Guayaquil in Ecuador (3ºS) to Cabo de Hornos (56ºS) in Chile [2], and up the south Atlantic coast (41ºS) of Argentina [3]. Recently, in a macro-scale work using microsatellite markers was proposed a biogeographical break in P. purpuratus ca. 40º South latitude, where two groups genetically divergent were identified northward and southward of the 40ºS [4]. Moreover, in phylogenetic studies using nuclear and mitochondrial markers, congruent phylogeographic patterns with such biogeographical break have also been observed [5, 6].
Morphological traits in sperm of P. purpuratus were compared and geographic intra-specific variation in spermatozoa traits of this mussel was observed [5], consistent with the phylogeographic patterns and genetic divergence previously reported. Sperm morphological traits have been used in systematic studies to differentiate closely related species, and in general sperm ultra-structure appears to be highly conserved at the species level [7, 8]. Accordingly, the sperm proteins play an important role in the process of speciation [9]. In this context, the recent developments in high-throughput sequencing techniques such as the whole transcriptome sequencing (RNAseq) have enabled large-scale analysis of genetic variation and gene expression in different tissues and species [10]. Furthermore, RNAseq is an effective way to discover genes participating in specific biological processes when genome reference or genome sequence is not available [11], as it is the P. purpuratus case. The aim of this study was to characterize the male gonad transcriptome of P. purpuratus. The information generated herein, may be used in future integrated analysis using transcriptome and proteomic data on this species and will allow the identification of a list of reproduction-related genes that could be useful to determine if populations of P. purpuratus are undergoing cryptic speciation mediated by reproductive isolation processes. Finally, our work represents the first transcriptomic resource for P. purpuratus and a reference transcriptome for closely related species.
For the characterization of the male gonad transcriptome, individuals of P. purpuratus were collected from the mid-intertidal zone off the coast of Valparaíso, Chile (32º57'S/71º33'W). In this species, sex can be easily determined based on gonad and mantle tissue colour, with male displaying a range of light colours (i.e. between white and yellow) and female showing browner coloration [3]. Gonadal tissue was obtained from mature males, preserved in RNAlater (Life Technologies), and stored at -80°C until use. To allow the optimization of RNA extraction, the total gonadal RNA for eight samples was extracted using the E.Z.N.A Total RNA Kit II (Omega Biotek), and then the sample of higher quality and concentration was selected for sequencing and library construction. mRNA enrichment and cDNA library construction were carried out using the Illumina Tru-SeqTM Stranded Total RNA Library Preparation Kit with Ribozero (human/mouse/rat). The cDNA was sequenced on an Illumina Hiseq 2500 using 100 bp paired-end reads. Sequencing was performed by STAB Vida (Lisboa, Portugal).
The de novo transcriptome assembly for the gonadal tissue in P. purpuratus was performed using Trinity (Release 2013-11-10) [12]. The adapters and low quality bases were trimmed using the tool Trimmomatic [13]; reads or bases with Q ≤ 30 and with less than 36 bp in length were removed. The obtained transcripts were then annotated using Blast searches on the Swiss-Prot database [14] using an e-value threshold of 1E-05. To perform the quantitative assessment of the assembly and completeness, the BUSCO software version 3.0.2 (Benchmarking Universal Single-Copy Orthologs) [15] was applied using default setting and the Metazoa gene set as a reference, which consists of 978 single-copy genes. Later, the transcripts were annotated to the three principal functional categories: biological process, molecular function and cellular components using Blast2GO [16] where both the second and third levels were annotated with GO terms [17].
Similar to the results of previous studies of molluscs and other invertebrates, total RNA extractions yielded a single non-degraded ribosomal RNA band that had the same size as that typical of 18S rRNA [18]. The co-migration of 28S and 18S rRNA fragments prevented us from using the RNA Integrity Number (RIN); thus only the absence of RNA degradation was considered [18].
A total of 126.38 million paired-end reads were obtained from transcriptome sequencing. The de novo Trinity assembly resulted in 105.42 million high-quality paired-end reads with a mean length of 96.01 bp. From this, a total of 385,288 transcripts and 314,741 different unigenes were generated. The transcripts had an average length of 430.5 bp and an N50 of 445 bp (Table 1), and GC content was 35%, congruent with other mussel assemblies where mantle tissue was used [19, 20]. The results of BUSCO analysis [15] showed that of the 978 metazoa BUSCOs, 89.7% (878) were complete, 60.94% (596) complete and single-copy, 28.8% (282) complete and duplicated, and 10.1% (99) were fragmented. Because the fraction of missing BUSCOs was quite low (0.1%), we concluded that the transcriptome of P. purpuratus was sufficiently complete and comprehensive for further analysis.
Summary statistics of the sequencing, de novo assembly, and annotated transcripts of gonadal tissue of Perumytilus purpuratus male.
| Total | |
|---|---|
| Obtained from sequencing | |
| Reads | 126,380,260 |
| Number of bases (Mbp) | 12,131 |
| Mean read length (bp) | 101 |
| After trimming for de novo assembly | |
| Reads | 105,419,693 |
| Total base pairs (Gb) | 12,130 |
| Mean read length (bp) | 96.01 |
| De novo assembly | |
| Transcripts | 385,288 |
| Shortest transcript length (bp) | 201 |
| Longest transcript length (bp) | 16,455 |
| Mean transcript length (bp) | 290 |
| Average length | 430.5 |
| Average GC (%) | 35 |
| N50 (bp) | 445 |
| Transcripts retained for annotation | |
| Transcripts SWP database | 43,756 |
| Transcripts GO database | 37,992 |
| Shortest transcript length (bp) | 201 |
| Longest transcript length (bp) | 16,455 |
| Mean transcript length (bp) | 327 |
| Average length | 521 |
| Average GC (%) | 35.5 |
Of the 385,288 transcripts, 43,756 (11.4%) were successfully annotated by Blast in the Swiss-Prot database [14], and 37,992 (9.9 %) transcripts were annotated to the functional GO terms in Blast2GO [16].
Even though a total of 385,288 transcripts were observed, a lower percentage of these (9.9%) were successfully annotated to the functional GO terms. Although this result seems to be quite low, a similar percentage of GO annotations have been reported in other non-model molluscan species, such as in pearl oysters [21] and the mussel Perna viridis [22]. The lower percentage of GO annotations is probably related to the fact that the number of results from the BLAST search for the de novo transcriptomes in a non-model species is highly dependent on the availability of the annotated sequence information for a reference genome [23, 24]. For instance, an increased percentage of GO annotations (> 25%) were shown in mollusc species with a large transcriptome characterization, as in the case of Haliotis species [25] and in mussels of the genus Mytilus [26].
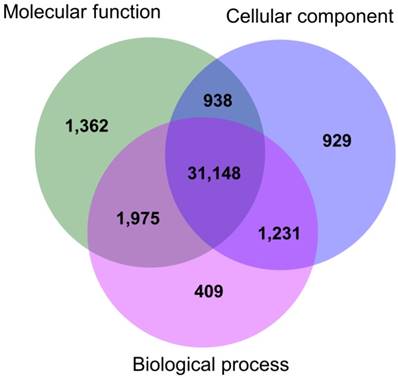
The functional annotations of the gene ontology (at second-level) showed that 34,763 transcripts were related to biological processes (i.e. 91.50% of the total GO annotated transcripts), 35,423 (93.24%) were involved in molecular functions, and 34,246 transcripts (90.14%) were associated with cellular components (Fig. 1). Among the total number of transcripts, 31,148 (81.99%) were associated to at least one of the three ontology categories; thus, most of the annotated genes were involved in multiple biological functions (Fig. 1). The functional descriptions of the basic GO categories, for the second and third-level, are shown in Fig. 2.
Venn diagram for number of transcripts annotated in each GO basic category: Biological process, Molecular function, and Cellular component.
Proportions of gene ontology annotations to gonadal tissue of Perumytilus purpuratus male. The functional descriptions for each basic GO category are shown: biological process (a), molecular function (b), and cellular components (c). GO second level is nested on GO third level.
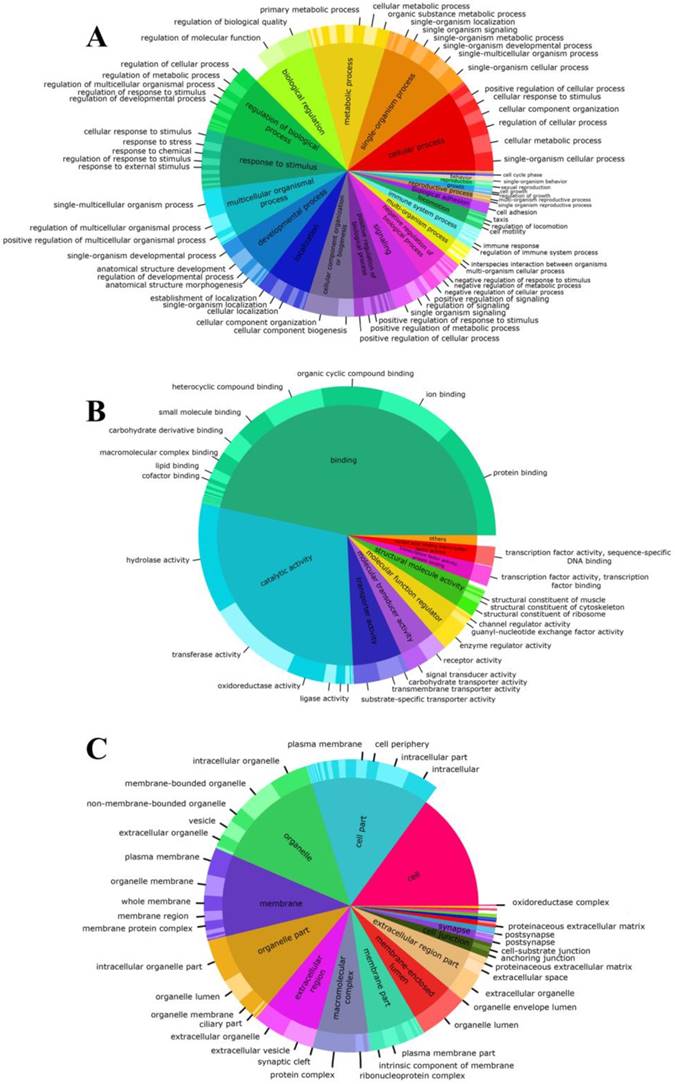
Representing the basic processes performed by the organism, the biological process was the most diverse category for both the second and third level, where the three principal GO terms were associated to cellular (95%), single organism (90%) and metabolic (83%) processes (Fig. 2A). Conversely, the molecular function category was the least variable (Fig. 2B) with transcripts that were associated principally to binding processes (93%) and catalytic processes (58%) terms. Among molecular function, we also found that in the third level, binding-related transcripts had different binding targets. Specifically, protein-binding term was associated with the highest number of transcripts (80%). Finally, the annotated transcripts associated with cellular components (Fig. 2C), indicate that the basic cellular terms were successfully identified. In this category, most transcripts were assigned to cell and organelle-related genes and were involved in some type of cellular structural organization. Details of the number of transcripts involved in the second and third-level for each GO category are shown in the Electronic supplementary material.
At last, our de novo transcriptome has shown a total of 5,075 transcripts related with reproductive process and reproduction terms in the biological process category, as for example: single organism reproductive, multi-organism reproductive, developmental process involved in reproduction and sexual reproduction (Fig. 2A and Electronic supplementary material).
In addition, a specific subset of GO annotations was generated using the “sperm” term. As result, a total of 1,294 sequences were assigned to the three functional GO categories, being the biological process category the more highly represented (1,002 sequences), followed by cellular component (282 sequences) and molecular function (10 sequences). In this search, many transcripts were related to sperm-specific processes. Accordingly, the transcripts from cellular component category were related to specific sperm structures as for instance, the acrosomal vesicle (GO: 0001669), sperm flagellum (GO: 0097223) and sperm midpiece (GO: 0097225). Whereas biological process transcripts were strongly associated to sperm-processes required for fertilization such as spermatogenesis (GO: 0007283), sperm capacitation (GO: 0048240), sperm motility (GO: 0030317) and sperm ejaculation (GO: 0042713), and also processes where the sperm-egg interaction could be implicated such as prevention of polyspermy (GO: 0060468), regulation of fusion of sperm to egg plasma membrane (GO: 0043012), fusion of sperm to egg plasma membrane (GO: 0007342), sperm-egg recognition (GO: 0035036) and binding of sperm to zona pellucida (GO: 0007339).
Gamete-recognition proteins, particularly in free-spawning marine invertebrates [27-29], have been widely studied. The evidence suggests that these proteins often evolve rapidly [30] and could have an important role on reproductive isolation during early stages of the speciation process [30]. Nevertheless, to identify more accurately gamete-recognition proteins, such as binding-sperm proteins, future experimental analyses should compare the gonad transcriptome expression profiles in males and females of P. purpuratus at different maturation stages. Finally, our de novo transcriptome and its functional annotations provide a valuable molecular resource to improve our knowledge of reproductive and fertilization-related genes in P. purpuratus.
Supplementary Material
Supplementary tables.
Acknowledgements
Financial support for this study was provided by the Fondo Nacional de Desarrollo Científico y Tecnológico, FONDECYT (Postdoctoral Project 3140250 to CB). The authors thank to the Instituto Español de Oceanografía (Vigo, Spain) and NGS team of StabVida (Portugal).
Accession numbers
Transcriptome data supporting is available in the NCBI Short Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra/) in the Bioproject PRJNA343253 under SRA accession number SRR4343820.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Guiñez R, Castilla JC. A tridimensional self-thinning model for multilayered intertidal mussels. Am Nat. 1999;154:341-57
2. Zagal C, Hermosilla C. Guía de invertebrados marinos del litoral Valdiviano. 1st edn, independent edition ed. Santiago de Chile, 217p.: Quebecor World Chile S.A. 2001
3. Osorio C, Bahamonde N. Moluscos bivalvos en pesquerías Chilenas. Biología Pesquera, Chile. 1968;3:69-128
4. Guiñez R, Pita A, Pérez M, Briones C, Navarrete SA, Toro J. et al. Present-day connectivity of historical stocks of the ecosystem engineer Perumytilus purpuratus along 4500 km of the Chilean Coast. Fish Res. 2016;7:322-32
5. Briones C, Guiñez R, Garrido O, Oyarzun PA, Toro JE, Pérez M. Sperm polymorphism and genetic divergence in the mussel Perumytilus purpuratus. Mar Biol. 2012;159:1865-70
6. Trovant B, Orensanz JML, Ruzzante DE, Stotz W, Basso NG. Scorched mussels (Bivalvia: Mytilidae: Brachidontinae) from the temperate coasts of South America: Phylogenetic relationships, trans-Pacific connections and the footprints of Quaternary glaciations. Mol Phylogenet Evol. 2015;82:60-74
7. Garrido O, Gallardo CS. Ultrastructure of sperms in bivalve molluscs of the Mytilidae family. Invertebr Reprod Dev. 1996;29:95-102
8. Introini GO, De Magalhaes CA, Aguiar-Jr O, Quaresma AJC, Lino-Neto J, Recco-Pimentel SM. Spermatozoan morphology of Brachidontes darwinianus and Brachidontes solisianus (Bivalvia, Mytilidae), from the southern Brazilian coast. Invertebr Reprod Dev. 2004;46:149-58
9. Swanson WJ, Vacquier VD. Reproductive protein evolution. Annu Rev Ecol Syst. 2002;33:161-79
10. Pareek CS, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Genet. 2011;52:413-35
11. Teaniniuraitemoana V, Huvet A, Levy P, Klopp C, Lhuillier E, Gaertner-Mazouni N. et al. Gonad transcriptome analysis of pearl oyster Pinctada margaritifera: identification of potential sex differentiation and sex determining genes. BMC Genomics. 2014;15:491
12. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644-52
13. Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20
14. Gasteiger E, Jung E, Bairoch A. Swiss-Prot: connecting biomolecular knowledge via a protein database. Mol Biol. 2001;3:47-55
15. Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210-2
16. Conesa A, Götz S, G-G JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674-6
17. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM. et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25-9
18. Dheilly NM, Lelong C, Huvet A, Favrel P. Development of a Pacific oyster (Crassostrea gigas) 31,918-feature microarray: identification of reference genes and tissue-enriched expression patterns. BMC Genomics. 2011;12:468
19. Yarra T, Gharbi K, Blaxter M, Peck LS, Clark MS. Characterization of the mantle transcriptome in bivalves: Pecten maximus, Mytilus edulis and Crassostrea gigas. Mar Genom. 2016;27:9-15
20. Bjarnmark NA, Yarra T, Churcher AM, Felix RC, Clark MS, Power DM. Transcriptomics provides insight into Mytilus galloprovincialis (Mollusca: Bivalvia) mantle function and its role in biomineralisation. Mar Genom. 2016;27:37-45
21. Huang X-D, Zhao M, Liu W-G, Guan Y-Y, Shi Y, Wang Q. et al. Gigabase-scale transcriptome analysis on four species of pearl oysters. Mar Biotechnol. 2013;15:253-64
22. Jiang X, Qiu LG, Zhao HW, Song QQ, Zhou HL, Han Q. et al. Transcriptomic responses of Perna viridis embryo to Benzo(a)pyrene exposure elucidated by RNA sequencing. Chemosphere. 2016;163:125-32
23. Leung PTY, Ip JCH, Mak SST, Qiu JW, Lam PKS, Wong CKC. et al. De novo transcriptome analysis of Perna viridis highlights tissue-specific patterns for environmental studies. BMC Genomics. BMC Genomics. 2014;15:804
24. Coppe A, Bortoluzzi S, Murari G, Marino IAM, Zane L, Papetti C. Sequencing and characterization of striped venus transcriptome expand resources for clam fishery genetics. Plos One. 2012;7:e44185
25. Harney E, Dubief B, Pierre Boudry P, Basuyaux O, Schilhabel MB, Huchette S. et al. De novo assembly and annotation of the European abalone Haliotis tuberculata transcriptome. Mar Genom. 2016 http://dx.doi.org/10.1016/j.margen.2016.03.002
26. Moreira R, Pereiro P, Canchaya C, Posada D, Figueras A, Novoa B. RNA-Seq in Mytilus galloprovincialis: comparative transcriptomics and expression profiles among different tissues. BMC Genomics. 2015;16:728
27. Springer SA, Crespi BJ. Adaptive gamete-recognition divergence in a hybridizing Mytilus population. Evolution. 2007;61:772-83
28. Galindo BE, Vacquier VD, Swanson WJ. Positive selection in the egg receptor for abalone sperm lysin. P Natl Acad Sci USA. 2003;100:4639-43
29. Zigler KS, Lessios HA. Evolution of bindin in the pantropical sea urchin Tripneustes: Comparisons to bindin of other genera. Mol Biol Evol. 2003;20:220-31
30. Vacquier VD, Swanson WJ. Selection in the rapid evolution of gamete recognition proteins in marine invertebrates. CSH Perspec Biol. 2011;3:a002931
Author contact
![]() Corresponding author: Dr. Carolina Briones Ortega <carolina.brionescl>
Corresponding author: Dr. Carolina Briones Ortega <carolina.brionescl>