ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2020; 8:1-6. doi:10.7150/jgen.40978 This volume Cite
Research Paper
Genomic Characteristics of the Toxic Bloom-Forming Cyanobacterium Microcystis aeruginosa NIES-102
Haruyo Yamaguchi1 ![]() , Shigekatsu Suzuki1, Yasunori Osana2, Masanobu Kawachi1
, Shigekatsu Suzuki1, Yasunori Osana2, Masanobu Kawachi1
1. Center for Environmental Biology and Ecosystem Studies, National Institute for Environmental Studies, 16-2 Onogawa, Tsukuba, Ibaraki 305-8506, Japan.
2. Department of Electrical and Electronics Engineering, University of the Ryukyus, 1 Senbaru, Nishihara-cho, Okinawa 903-0213, Japan.
Received 2019-10-7; Accepted 2019-11-20; Published 2020-1-1
Abstract

Microcystis aeruginosa, a bloom-forming cyanobacterium distributed mainly in freshwater environments, can be divided into at least 12 groups (A-K and X) based on multi-locus phylogenetic analyses. In this study, we characterized the genome of microcystin-producing M. aeruginosa NIES-102, assigned to group A, isolated from Lake Kasumigaura, Japan. The complete genome sequence of M. aeruginosa NIES-102 comprised a 5.87-Mbp circular chromosome containing 5,330 coding sequences. The genome was the largest among all sequenced genomes for the species. In a comparison with the genome of M. aeruginosa NIES-843, which belongs to the same group, the microcystin biosynthetic gene cluster and CRISPR-Cas locus were highly similar. A synteny analysis revealed small-scale rearrangements between the two genomes. Genes encoding transposases were more abundant in these two genomes than in other Microcystis genomes. Our results improve our understanding of structural genomic changes and adaptation to a changing environment in the species.
Keywords: algal bloom, cyanobacteria, genome, microcystin, Microcystis aeruginosa, Lake Kasumigaura
Introduction
Toxic cyanobacterial blooms commonly occur in freshwater environments worldwide. During the summer, these blooms result in serious environmental problems, such as bad odors, bottom-layer anoxia, and cyanotoxin production. Microcystis aeruginosa is a unicellular, colony-forming cyanobacterium distributed primarily in eutrophic freshwater environments [1]. It is the most well-known toxic bloom-forming cyanobacteria; some strains produce hepatotoxic cyanotoxins called microcystins, which are the only cyanotoxins for which the World Health Organization has established guideline values for drinking water [2]. Global climate change, including global warming, is expected to increase the frequency of Microcystis blooms [1]. Microcystis has been a focus of research related to global climate change and the eutrophication of freshwater lakes.
Tanabe et al. classified M. aeruginosa isolates by a multi-locus phylogenetic analysis based on seven housekeeping genes and showed that the species has high intraspecific genetic diversity [3]. Using this approach, M. aeruginosa isolates can be divided into at least 12 phylogenetic groups (A-K and X). The strains in groups A and X and some strains in group B produce microcystins [3, 4].
To date, 4 complete, 22 scaffold-level, and 31 contig-level genome sequences of M. aeruginosa have been registered in the National Center for Biotechnology Information Genome database (https://www.ncbi.nlm.nih.gov/genome/genomes/820). M. aeruginosa NIES-87, 98, 298, 843, 2481, and 2549 were isolated from a shallow, hyper-eutrophic lake, Lake Kasumigaura, in Japan [5-10], where algal blooms occur every summer to fall [11]. M. aeruginosa in Lake Kasumigaura has high genetic diversity [12], emphasizing the important of additional sequence information for strains in the lake.
Microcystis aeruginosa NIES-102 was collected from Lake Kasumigaura in 1982. A previous phylogenetic analysis has shown that this strain belongs to group A [12]. M. aeruginosa NIES-102 is of particular interest owing to its production of microcystins, mainly microcystin RR [13]. In addition, microviridin, a protease inhibitor produced by several cyanobacteria, was first discovered in this strain [14]. In this study, we report the complete genome sequence of M. aeruginosa NIES-102 and the results of a comparative genomic analysis with other M. aeruginosa genomes.
Materials and Methods
An axenic culture of M. aeruginosa NIES-102 was obtained from the Microbial Culture Collection at the National Institute for Environmental Studies, Japan (http://mcc.nies.go.jp/). DNA extraction from a 20 mL culture of M. aeruginosa NIES-102 was performed using NucleoBond Buffer Set III and NucleoBond AXG 100 (Macherey-Nagel, Düren, Germany), following the manufacturer's instructions. DNA sequencing was performed using a MinION sequencer (Oxford Nanopore Technologies, Oxford, UK) and Illumina MiSeq (San Diego, CA, USA). For MinION sequencing, a DNA library was prepared using the Rapid Sequencing Kit (SQK-RAD001) following standard protocols provided by Oxford Nanopore Technologies. The MinION MK1 sequencer and flow cell (R9.4.1) were used for sequencing. In total, 118,979 reads (656,208,396 bp) were obtained. For Illumina MiSeq sequencing, DNA was fragmented using the Covaris M220 Ultrasonicator (Woburn, MA, USA) to obtain 550-bp reads. The DNA library was prepared using the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA) following the manufacturer's protocol. Sequencing was performed using the 600-cycle MiSeq Reagent Kit v.3. In total, 1,742,106 paired-end reads (949,209,678 bp in total) were obtained. Error correction for nanopore reads was performed using Nanocorr 0.01 [15]. The corrected nanopore reads were assembled into a single contig using Canu v.1.5 [16]. The corrected reads were aligned to the contig using BWA-MEM 0.7.17 with a default option [17]. The contig was polished using Pilon 1.22 [18]. The genome was annotated using DFAST [19] with CyanoBase [20] as organism-specific database. A chromosome map of this strain was drawn using DNAPlotter [21]. Secondary metabolites were predicted using antiSMASH [22] with default settings. Clustered regularly interspaced short palindromic repeat (CRISPR) loci were detected using CRISPRCasFinder [23]. Furthermore, cas genes were identified using eggNOG-mapper v.2 [24] and BLASTP [25]. Functional annotation was performed using eggNOG-mapper v.2 [24]. Synteny was analyzed using Murasaki [26]. The localization of transposases was evaluated using CGView [27].
Results and Discussion
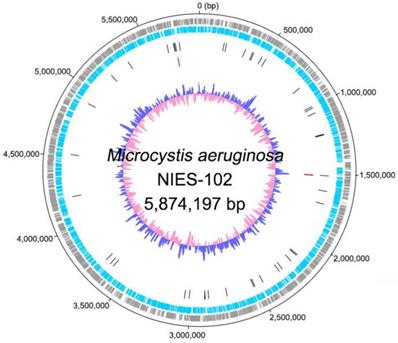
Genomic characteristics of Microcystis aeruginosa NIES-102 are summarized in Table 1. We obtained a genome consisting of a 5.87-Mbp circular chromosome (Fig. 1). Nanopore MinION and Illumina MiSeq read coverages were 112-fold and 162-fold, respectively. The genome of M. aeruginosa NIES-102 was the largest among complete genomes of M. aeruginosa. It included 5,330 protein-coding sequences, 44 tRNA genes, and two sets of rRNA genes. The G+C content was 42.39%. As the result of GC skew analysis, origin of the replication could not be identified. Using antiSMASH 5.0.0 for prediction, we identified 11 secondary metabolite gene clusters, including microcystin [28], microviridin B [29], aeruginosin [30], and micropeptin biosynthetic gene clusters [31]. CRISPRCasFinder predicted a single CRISPR-Cas locus with strong support in the genome with a length of 3,437 bp. The consensus CRISPR repeat sequence was 5′-GTTCCAATTAATCTTAAACCCTATTAGGGATTGAAAC-3′ (37 bp) and there were 47 spacers. According to an established classification system for CRISPER-Cas [32], the locus was subtype I-D CRISPR-Cas 2, consisting of eight genes (cas3, csc3/cas10d, csc2, csc1, cas6, cas4, cas1, and cas2).
General characteristics of M. aeruginosa NIES-102 and NIES-843
| Features | NIES-102 (this study) | NIES-843 (Kaneko et al. 2008) |
|---|---|---|
| Genome size (bp) | 5,874,197 | 5,842,795 |
| G+C content (%) | 42.39 | 42.33 |
| Coding sequence (CDS) | 5,330 | 5,897 |
| rRNA operon | 2 | 2 |
| tRNA genes | 44 | 42 |
| Locality | Lake Kasumigaura, Japan | Lake Kasumigaura, Japan |
| Date of collection | Sep. 1982 | Aug. 1997 |
Complete chromosome map of Microcystis aeruginosa NIES-102. The chromosome map comprises five concentric circles. The gray and light-blue circles show the positions of protein-coding genes on the plus and minus strands, respectively. Black bars on the third circle, red bars on the fourth circle, and blue/pink circle show tRNA, rRNA genes, and guanine-cytosine content.
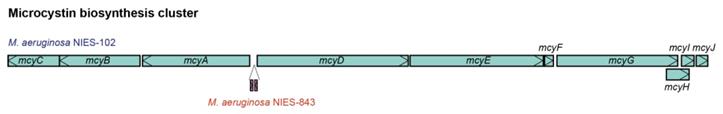
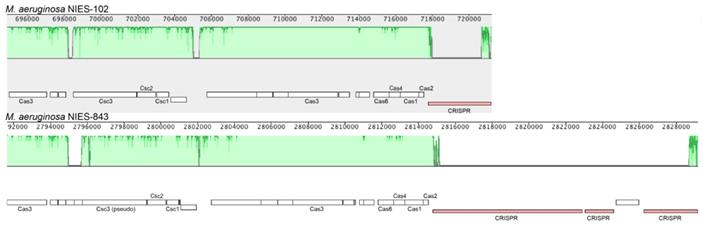
We compared the genome of M. aeruginosa NIES-102 with those of other M. aeruginosa strains. The genomes of M. aeruginosa NIES-102 and M. aeruginosa NIES-843 (group A) shared similar sizes as well as numbers and kinds of genes (Table 1 and 2). The genomes both possess two rRNA operons and the 16S rRNA gene sequences shared 99.7% similarity (5/1485 bp differences). The two strains had similar microcystin biosynthetic gene clusters (Fig. 2); however, two hypothetical proteins were inserted between mcyA and mcyD in M. aeruginosa NIES-843. The similarity of mcy genes between M. aeruginosa NIES-102 and NIES-843 were 99% excluding mcyF, mcyH, mcyJ (100%) and mcyD (98%). Four types of CRISPR-Cas systems have been reported in M. aeruginosa [32]. The CRISPR-Cas locus in each strain was classified as subtype I-D. However, the numbers and positions of genes in the CRISPR-associated gene clusters differed between the two strains (Fig. 3). These results suggested that the M. aeruginosa NIES-102 genome has similar characteristics to those of the M. aeruginosa NIES-843 genome, reflecting their close phylogenetic relationship [3].
Clusters of orthologous group categories of M. aeruginosa NIES-102 and NIES-843
| Category | Definition | NIES-102 | NIES-843 |
|---|---|---|---|
| Cellular processes and signaling | |||
| D | Cell cycle control, cell division, chromosome partitioning | 84 | 88 |
| M | Cell wall/membrane/envelope biogenesis | 228 | 226 |
| N | Cell motility | 70 | 79 |
| O | Post-translational modification, protein turnover, and chaperones | 184 | 181 |
| T | Signal transduction mechanisms | 178 | 177 |
| U | Intracellular trafficking, secretion, and vesicular transport | 82 | 85 |
| V | Defense mechanisms | 72 | 72 |
| W | Extracellular structures | 1 | 1 |
| Z | Cytoskeleton | 1 | 0 |
| Information storage and processing | |||
| A | RNA processing and modification | 3 | 6 |
| B | Chromatin structure and dynamics | 1 | 1 |
| J | Translation, ribosomal structure and biogenesis | 197 | 198 |
| K | Transcription | 195 | 199 |
| L | Replication, recombination and repair | 828 | 880 |
| Metabolism | |||
| C | Energy production and conversion | 227 | 228 |
| E | Amino acid transport and metabolism | 210 | 207 |
| F | Nucleotide transport and metabolism | 99 | 99 |
| G | Carbohydrate transport and metabolism | 127 | 132 |
| H | Coenzyme transport and metabolism | 175 | 173 |
| I | Lipid transport and metabolism | 82 | 91 |
| P | Inorganic ion transport and metabolism | 183 | 185 |
| Q | Secondary metabolites biosynthesis, transport, and catabolism | 110 | 120 |
| Poorly characterized | |||
| S | Function unknown | 1314 | 1272 |
Comparison of microcystin biosynthesis clusters between M. aeruginosa NIES-102 and NIES-843. The microcystin biosynthesis cluster of M. aeruginosa NIES-843 differs from that of NIES-102 in having two additional genes between mcyA and mcyD.
Comparison of the CRISPR-Cas locus between M. aeruginosa NIES-102 and NIES-843. The genomes of M. aeruginosa NIES-102 and NIES-843 have a subtype I-D CRISPR-Cas locus, although the numbers and positions of inserted genes in CRISPR associated genes differ between the two genomes. Only CRISPR-Cas related genes and CRISPR are indicated. The figure was drawn using Mauve software (http://darlinglab.org/mauve/mauve.html).
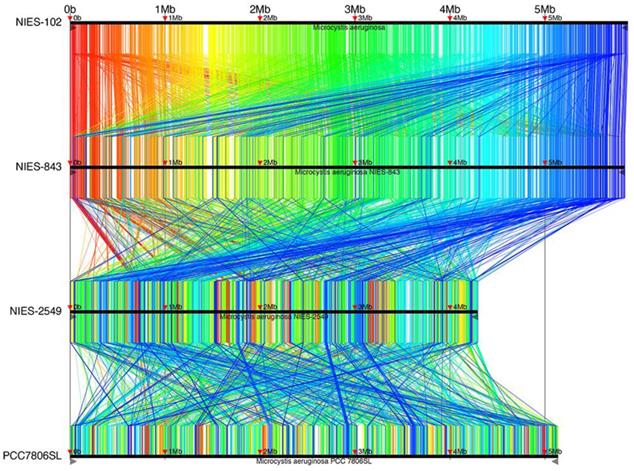
Complete genomes of M. aeruginosa have been reported for strains NIES-2481, NIES-2549, and PCC7806SL [33] in addition to NIES-843; M. aeruginosa NIES-2481 and NIES-2549 are assigned to group G, but M. aeruginosa PCC7806SL is unclassified. To identify genomic rearrangements, we conducted a synteny analysis using these strains (Fig. 4). 9,806 conserved regions of length 34-6,489 bp are shown in Fig. 4. The results are filtered by tf-idf scoring feature of Murasaki to remove sequences of high occurrence frequency such as repeat sequences: every region is expected to be highly specific even if the length is as short as 34 bp. The general genomic structures of M. aeruginosa NIES-102 and NIES-843 were conserved, with small rearrangements scattered throughout. This result also supports the close relationship between these two strains. We detected frequent recombination between M. aeruginosa NIES-843 and NIES-2549 and between M. aeruginosa NIES-2549 and PCC7806SL, suggesting substantial divergence between these strains. These results revealed high genomic plasticity in M. aeruginosa.
Synteny analysis of M. aeruginosa NIES-102, NIES-843, NIES-2549 and PCC 7806SL. Similar genomic regions in the four genomes are indicated with the same colors and lines.
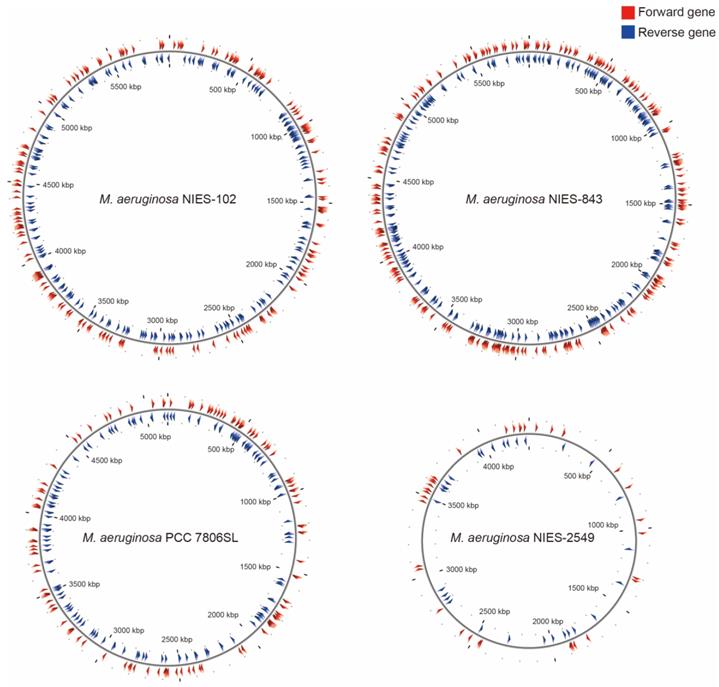
Among M. aeruginosa strains with complete genomes, M. aeruginosa NIES-102 (5.8 Mb) had the largest genome and M. aeruginosa NIES-2549 (4.3 Mb) had the smallest genome. The species clearly exhibits diversity in genome size. Yamaguchi et al. [9] suggested that the genome size difference between group A (NIES-843) and group G (NIES-2549) can be partly explained by a difference in the number of genes involved in replication, recombination, and repair (category L, COG). We performed functional annotation using eggNOG-mapper v. 2 against M. aeruginosa NIES-102 and NIES-843 genomes (Table 2). The number of orthologous groups assigned to category L in M. aeruginosa NIES-102 was similar to that in M. aeruginosa NIES-843, suggesting that strains in group A share a large number of genes in category L. Within category L, transposases contribute substantially to variation in genome size. Humbert et al. (2013) showed that the M. aeruginosa genome includes a high proportion of genes encoding transposases, providing a basis for rapid divergence and survival in harsh freshwater environments [34]. We found that the transposase-coding genes in M. aeruginosa NIES-102 and NIES-843 were scattered at a high density throughout the genomes (Fig. 5). In M. aeruginosa PCC7806SL, the density of transposases was lower than those in M. aeruginosa NIES-102 and NIES-843. M. aeruginosa NIES-2549 had the fewest transposases among the four genomes. We detected far more genes encoding transposases in group A than in group G, and these genes may contribute to expansions and contractions of M. aeruginosa genomes. Additional genomic analyses are needed to explain the high number of transposes in group A.
Localizations of transposases in M. aeruginosa NIES-102, NIES-843, NIES-2549 and PCC 7806SL. Red arrowheads indicate forward genes, and blue arrowheads indicate reverse genes.
In Japanese lakes, including Lake Kasumigaura, M. aeruginosa group A is frequently observed [12]. The high frequency of strains in group A may be explained by the abundance of genes related to environmental adaptation, such as transposases, in this group. Since freshwater environments change drastically, these genes may promote survival. Climate change and global warming are expected to result in frequent occurrences of algal blooms. Additional genomic information for M. aeruginosa would improve our understanding and management of freshwater ecosystems in Japan.
Acknowledgements
This study was partially supported by the National Bioresource Project Algae of the Japan Agency for Medical Research and Development (AMED).
Accession Numbers
The whole genome shotgun project for M. aeruginosa NIES-102 has been deposited in DDBJ under accession no. AP019314.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Harke MJ, Steffen MM, Gobler CJ. et al. A review of the global ecology, genomics, and biogeography of the toxic cyanobacterium, Microcystis spp. Harmful Algae. 2016;54:4-20
2. World Health Organization. Guidelines for Drinking-Water Quality. Fourth Edition Incorporating the First Addendum. Geneva, Switzerland: World Health Organization. 2017
3. Tanabe Y, Kasai F, Watanabe MM. Multilocus sequence typing (MLST) reveals high genetic diversity and clonal population structure of the toxic cyanobacterium Microcystis aeruginosa. Microbiology. 2007;153:3695-3703
4. Tanabe Y, Hodoki Y, Sano T. et al. Adaptation of the freshwater bloom-forming cyanobacterium Microcystis aeruginosa to brackish water is driven by recent horizontal transfer of sucrose genes. Front Microbiol. 2018;9:1150
5. Yamaguchi H, Suzuki S, Kawachi M. Draft genome sequence of Microcystis aeruginosa NIES-87, a bloom-forming cyanobacterium from Lake Kasumigaura, Japan. Genome Announc. 2018;6:e01596-17
6. Yamaguchi H, Suzuki S, Sano T. et al. Draft genome sequence of Microcystis aeruginosa NIES-98, a non-microcystin-producing cyanobacterium from Lake Kasumigaura, Japan. Genome Announc. 2016;4:e01187-16
7. Yamaguchi H, Suzuki S, Kawachi M. Improved draft genome sequence of Microcystis aeruginosa NIES-298, a microcystin-producing cyanobacterium from Lake Kasumigaura, Japan. Genome Announc. 2018;6:e01551-17
8. Kaneko T, Nakajima N, Okamoto S. et al. Complete genomic structure of the bloom-forming toxic cyanobacterium Microcystis aeruginosa NIES-843. DNA res. 2007;14:247-256
9. Yamaguchi H, Suzuki S, Osana Y. et al. Complete genome sequence of Microcystis aeruginosa NIES-2481 and common genomic features of group G M. aeruginosa. J Genomics. 2018;6:30-33
10. Yamaguchi H, Suzuki S, Tanabe Y. et al. Complete genome sequence of Microcystis aeruginosa NIES-2549, a bloom-forming cyanobacterium from Lake Kasumigaura, Japan. Genome Announc. 2015;3:e00551-15
11. Takamura N, Watanabe MM. Seasonal changes in the biomass of four species of Microcystis in Lake Kasumigaura. Japanese Journal of Limnology. 1987;48:139-144
12. Tanabe Y, Watanabe MM. Local expansion of a panmictic lineage of water bloom-forming cyanobacterium Microcystis aeruginosa. PLoS One. 2011;6:e17085
13. Kaya K, Watanabe MM. Microcystin composition of an axenic clonal strain of Microcystis viridis and Microcystis viridis-containing waterblooms in Japanese freshwaters. J Appl Phycol. 1990;2:173-178
14. Ishitsuka MO, Kusumi T, Kakisawa H. et al. Microviridin. A novel tricyclic depsipeptide from the toxic cyanobacterium Microcystis viridis. J Am Chem Soc. 1990;112:8180-8182
15. Goodwin S, Gurtowski J, Ethe-Sayers S. et al. Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome research. 2015;25:1750-1756
16. Koren S, Walenz BP, Berlin K. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome research. 2017;27:722-736
17. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013:1303 3997
18. Walker BJ, Abeel T, Shea T. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one. 2014;9:e112963
19. Tanizawa Y, Fujisawa T, Nakamura Y. DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics. 2017;34:1037-1039
20. Fujisawa T, Narikawa R, Maeda SI. et al. CyanoBase: a large-scale update on its 20th anniversary. Nucleic Acids Res. 2017;45:D551-D554
21. Carver T, Thomson N, Bleasby A. et al. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2008;25:119-120
22. Weber T, Blin K, Duddela S. et al. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237-W243
23. Couvin D, Bernheim A, Toffano-Nioche C. et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic acids res. 2018;46:W246-W251
24. Huerta-Cepas J, Forslund K, Coelho LP. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol Evol. 2017;34:2115-2122
25. Gish W, States DJ. Identification of protein coding regions by database similarity search. Nature genetics. 1993;3:266-272
26. Popendorf K, Tsuyoshi H. Osana Y. et al. Murasaki: a fast, parallelizable algorithm to find anchors from multiple genomes. PLoS One. 2010;5:e12651
27. Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics. 2004;21:537-539
28. Tillett D, Dittmann E, Erhard M. et al. Structural organization of microcystin biosynthesis in Microcystis aeruginosa PCC7806: an integrated peptide-polyketide synthetase system. Chemistry & biology. 2000;7:753-764
29. Ziemert N, Ishida K, Liaimer A. et al. Ribosomal synthesis of tricyclic depsipeptides in bloom-forming cyanobacteria. Angewandte Chemie International Edition. 2008;47:7756-7759
30. Ishida K, Welker M, Christiansen G. et al. Plasticity and evolution of aeruginosin biosynthesis in cyanobacteria. Appl Environ Microbial. 2009;75:2017-2026
31. Nishizawa T, Ueda A, Nakano T. et al. Characterization of the locus of genes encoding enzymes producing heptadepsipeptide micropeptin in the unicellular cyanobacterium Microcystis. J Biochem. 2011;149:475-485
32. Yang C, Lin F, Li Q. et al. Comparative genomics reveals diversified CRISPR-Cas systems of globally distributed Microcystis aeruginosa, a freshwater bloom-forming cyanobacterium. Front Microbiol. 2015;6:394
33. Zhao L, Song Y, Li L. et al. The highly heterogeneous methylated genomes and diverse restriction-modification systems of bloom-forming Microcystis. Harmful algae. 2018;75:87-93
34. Humbert JF, Barbe V, Latifi A. et al. A tribute to disorder in the genome of the bloom-forming freshwater cyanobacterium Microcystis aeruginosa. PLoS One. 2013;8:e70747
Author contact
![]() Corresponding author: Haruyo Yamaguchi, Center for Environmental Biology and Ecosystem Studies, National Institute for Environmental Studies, 16-2 Onogawa, Tsukuba, Ibaraki 305-8506, Japan; Tel.: +81-29-850-2424; Fax: +81-29-850-2587; E-mail: yamaguchi.haruyogo.jp.
Corresponding author: Haruyo Yamaguchi, Center for Environmental Biology and Ecosystem Studies, National Institute for Environmental Studies, 16-2 Onogawa, Tsukuba, Ibaraki 305-8506, Japan; Tel.: +81-29-850-2424; Fax: +81-29-850-2587; E-mail: yamaguchi.haruyogo.jp.