ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2020; 8:37-42. doi:10.7150/jgen.38516 This volume Cite
Research Paper
Draft Genome Sequences of Bifidobacterium animalis Consecutively Isolated from Healthy Japanese Individuals
Tomoya Tsukimi1,2, Tsubasa Watabe1,2, Kazuki Tanaka1,2, Mitsuhiko P. Sato3, Haruo Suzuki1,4, Masaru Tomita1,2,4, Shinji Fukuda1,2,5,6,7 ![]()
1. Institute for Advanced Biosciences, Keio University, 246-2 Mizukami, Kakuganji, Tsuruoka, Yamagata 997-0052, Japan.
2. Systems Biology Program, Graduate School of Media and Governance, Keio University, 5322 Endo, Fujisawa, Kanagawa 252-0882, Japan.
3. Department of Bacteriology, Faculty of Medical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi, Fukuoka 812-8582, Japan.
4. Faculty of Environment and Information Studies, Keio University, 5322 Endo, Fujisawa, Kanagawa 252-0882, Japan.
5. Intestinal Microbiota Project, Kanagawa Institute of Industrial Science and Technology, 3-25-13 Tonomachi, Kawasaki, Kanagawa 210-0821, Japan.
6. Transborder Medical Research Center, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8575, Japan.
7. Metabologenomics, Inc., 246-2 Mizukami, Kakuganji, Tsuruoka, Yamagata 997-0052, Japan.
Received 2019-7-17; Accepted 2019-11-20; Published 2020-4-6
Abstract

Bifidobacterium species are well recognized as probiotics and colonized in various parts of the human body. Here, we report the draft genome sequences of Bifidobacterium animalis isolated from two healthy Japanese volunteers, one of which was sampled twice before and after a 10-year interval. A core genome phylogeny analysis indicated that the strains isolated from the same volunteer were closely related. This paper is the first report of multiple draft genome sequences of B. animalis independently isolated from the same individual and provides insight into the probiotic potential of a member of this species.
Keywords: Whole-Genome Sequencing, Intestinal Microbiota, Bifidobacterium animalis
Introduction
About 40 trillion of bacteria inhabit the human body, and the majority of these are located in the gastrointestinal tract [1]. Previous studies have shown a relationship between the human gut microbiota and various diseases [2,3], and some bacteria, which are referred to as probiotics, are considered to be beneficial for human health [4]. The genus Bifidobacterium is one well-known group of probiotic bacteria. Species belonging to the genus are gram-positive, rod-shaped, and are anaerobic [5]. They colonize in food, such as yogurt, and various parts of the human body including the gut, vagina, oral cavity, and breast milk [5]. In the human gut, they ferment carbohydrates into short-chain fatty acids (SCFAs) such as acetate, which have beneficial effects on human health [2]. Bifidobacterium species do not produce butyrate directly; however, they increase butyrate production via cross-feeding with butyrate-producing bacteria in the colon [6]. Bifidobacterium species have great potential as a probiotics, but they are not always colonized when they are supplied as a probiotic intervention [7]. The genus Bifidobacterium contains 69 species and 10 subspecies (http://www.bacterio.net/; accessed 26 February 2019) [8]. In this study, we focused on Bifidobacterium animalis, which has two subspecies, subsp. animalis and subsp. lactis. A previous study reported the beneficial effect of the species on ameliorating the risk of food allergy [9] and maintaning remission in ulcerative colitis [10]. Here, we report three draft genome sequences of B. animalis subsp. lactis isolated from two healthy Japanese individuals, one of which was sampled twice before and after a 10-year interval.
Materials and Methods
Sample Collection and Isolation of B. animalis
Fecal samples were collected from two healthy Japanese volunteers (one subject was 30 years old, and another was 35 and 45 years old at the time of sampling; Table 1) and stored at -80 °C until use.
Metadata of subjects in this study.
| Subject | Age at time of sampling | Gender | Intake frequency of yogurt | Last intake yogurt before fecal sampling | Strains contained in yogurt |
|---|---|---|---|---|---|
| 1 | 30 | Male | 3 or 4 times a week | Bifidus Yogurt* | Bifidobacterium longum, Lactobacillus delbrueckii subsp. bulgaricus, Streptococcus thermophilus |
| 2 | 35, 45 | Male | a few times a month | not applicable | not applicable |
*Bifidus Yogurt is commercial products of MORINAGA MILK INDUSTRY CO., LTD.
Each fecal sample (20 mg) was suspended in 1 mL of sterile phosphate-buffered saline (PBS), which was then used to generate two dilutions in PBS, 1:1,000 and 1:10,000. A 50 µL of aliquot from each suspension was inoculated onto a transgalactosylated oligosaccharide (TOS) propionate agar medium plate (Yakult Pharmaceutical Industry Co., Ltd., Tokyo, Japan), which can isolate Bifidobacterium from feces [11], and cultured for 1 day at 37 °C under anaerobic conditions. Ten colonies for each subject were randomly selected; inoculated with sterile pipette tips into a 96-well plate (Greiner Bio-One International GmbH, Frickenhausen, Germany) containing 500 µL of De Man, Rogosa and Sharpe (MRS) liquid medium (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan); and cultured for 1 day at 37 °C under anaerobic conditions.
After cultivation, we confirmed the identity of this species with PCR based on 16S rRNA gene amplification using a Bifidobacterium-specific primer [12]. Genomic DNA from samples confirmed as Bifidobacterium was extracted with the DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany).
Genome Sequencing, Assembly, and Annotation
The sequencing libraries were prepared using the NEBNext Ultra II FS DNA Library Prep (New England Biolabs, Ipswich, Massachusetts, U.S.) and sequenced with the Illumina HiSeq 2500 (Illumina, Inc. San Diego, California, U.S.). In total 3,282,698 reads (which consisted of 151-bp paired-end reads) were produced (1-Y0; 1,221,120 reads, 2-Y0; 971,770 reads, 2-Y10; 1,089,808 reads). The reads were filtered for read quality using Platanus_trim v1.0.7 (http://platanus.bio.titech.ac.jp/pltanus_trim; accessed 23 February 2019) with default parameters, which resulted in the retention of 3,265,842 reads (1-Y0; 1,215,194 reads, 2-Y0; 965,958 reads, 2-Y10; 1,084,690 reads). Trimmed reads were assembled with Platanus v1.2.4 [13] with default parameters, and contigs that were ≤ 300 bp were discarded based on an in-house script deposited in GitHub (https://github.com/MitsuhikoP/cut_short_fasta.git). Genome completeness was estimated using Benchmarking Universal Single-Copy Orthologs (BUSCO) v1 [14] with the bacteria data set by submitting assembled sequences (FASTA formatted text file) as query to the gVolante web server [15] and Quality assessment tool for genome assemblies (QUAST) [16]. Of the 40 BUSCO group genes, all genes were completely or partially recovered in the assembly and the genomes fraction of three draft genome sequences to complete genome sequence of B. animalis subsp. lactis DSM 10140 were over 98 % (Table 2). Genes were annotated using the DDBJ Fast Annotation and Submission Tool (DFAST) pipeline [17]. Identification of B. animalis was performed with BLAST+ v2.4.0 [18,19], and pyani v0.2.7 (https://github.com/widdowquinn/pyani; accessed 23 February 2019), which is Python module to calculate average nucleotide identity (ANI). For comparative analysis, complete genome sequences of bacteria in GenBank format of B. animalis subsp. animalis ATCC 25527 (GCA_000260715.1_ASM26071v1) and subsp. lactis DSM 10140 (GCA_000022965.1_ASM2296v1) were downloaded using the command line tool wget from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/) on 27 March 2019.
Genomic features of samples used in this study.
| Sample | % BUSCO Score (Complete + Partial) | Genome fraction (%) | GC content (%) | Number of contigs | Total contig size (bp) | Largest contig (bp) | N50 (bp) | Number of CDSs |
|---|---|---|---|---|---|---|---|---|
| 1-Y0 | 100 | 98.8 | 60.5 | 15 | 1,917,421 | 681,743 | 339,970 | 1,571 |
| 2-Y0 | 100 | 98.8 | 60.5 | 15 | 1,918,142 | 1,021,733 | 1,021,733 | 1,561 |
| 2-Y10 | 100 | 98.4 | 60.5 | 15 | 1,910,072 | 681,755 | 339,940 | 1,561 |
Sample name represents the subject and the year (Y) after first sampling.
GC content (%): The relative frequency (percentage) of guanine and cytosine residues (G + C)/(A + T + G + C)
CDSs: coding sequences
Nucleotide sequence alignments for core genes, which are defined as single-copy genes that are common across all genomes, were produced using Roary v3.12.0 [20] with a minimum blastp percentage identity of 95, and MAFFT v7.407 [21]. A phylogenetic tree was constructed using FastTree v2.1.3 [22] with the GTR + CAT model. The phylogenetic tree was rooted with midpoint rooting using the phangorn package v2.4.0 (https://github.com/KlausVigo/phangorn) and drawn using the ape package v5.3 (http://ape-package.ird.fr/) in R v3.4.2 (https://www.r-project.org/). For SNP analysis the nucleotide sequences of 11 genomes of B. animalis subsp. lactis strain: DSM 10140 (GCA_000022965.1_ASM2296v1), AD011 (GCA_000021425.1_ASM2142v1), B420 (GCA_000277325.1_ASM27732v1), BB-12 (GCA_000025245.1_ASM2524v1), BLC1 (GCA_000224965.2_ASM22496v2), Bi-07 (GCA_000277345.1_ASM27734v1), Bl-04 (GCA_000022705.1_ASM2270v1), Bl12 (GCA_000414215.1_ASM41421v1), CNCM I-2494 (GCA_000220885.1_ASM22088v1), KLDS2.0603 (GCA_000816205.1_ASM81620v1) and V9 (GCA_000092765.1_ASM9276v1) were downloaded using the command line tool wget from the NCBI FTP site on 26 September 2019. The genome alignment of two genomes isolated from volunteer 2 and 11 downloaded genomes and calculating SNPs were performed using MAUVE v20150226 [23] with default parameters. Heatmap of SNP matrix was drawn using the corrplot package v0.84 (https://cran.r-project.org/web/packages/corrplot/) in R v3.4.2.
Results and Discussion
The three draft genome sequences of B. animalis analyzed in this study contained 15 contigs consisting of 1,910,072 to 1,918,142 bp, with a G+C content of 60.5% and 1,561 to 1,571 putative coding sequences (Table 2). The complete genome sequence of B. animalis is ~1.94 Mbp with a G+C content of 60.5 %, and 1,600 protein-encoding genes [24], which is most consistent with the sequence results of this study.
To infer the phylogenetic relationships, we built a rooted phylogenetic tree based on a concatenated nucleotide sequence alignment of the 1,176 core genes from 5 strains, included the 3 strains isolated from the volunteers and the two reference genomes of two B. animalis subspecies obtained by downloading from NCBI FTP site (Figure 1). The core genome phylogeny indicated that the most basal lineage was B. animalis subsp. animalis, which was followed by the clade consisting of B. animalis subsp. lactis, and the strains isolated from the volunteers.
Rooted phylogenetic tree of strains analyzed in this study. Rooted phylogenetic tree obtained from a concatenated nucleotide sequence alignment of the 1,176 core genes of five B. animalis strains, consisting of the three strains isolated from the two volunteers and reference genomes of two B. animalis subspecies. The horizontal bar at the base of the figure represents 0.001 substitutions per nucleotide site. The FastTree branch support values are indicated. 1-Y0, B. animalis isolate from volunteer 1; 2-Y0, B. animalis isolate from volunteer 2, first collection time point; 2-Y10, B. animalis isolate from volunteer 2, second collection time point
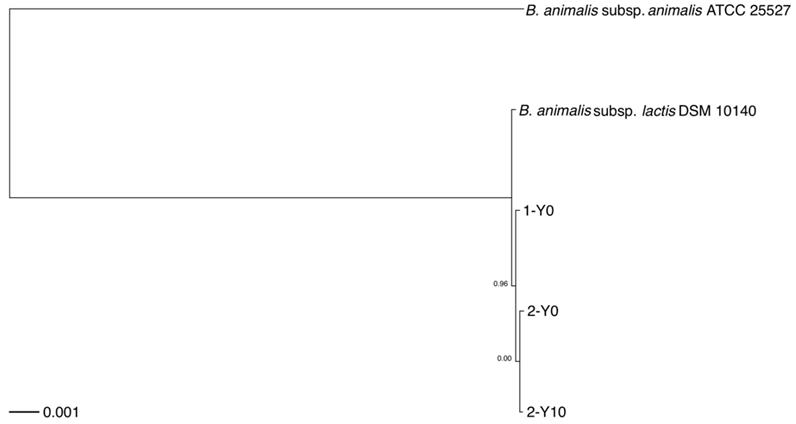
A previous study showed that B. animalis subsp. lactis was rarely found in intestinal biopsy samples, whereas it was frequently detected in fecal samples [25], indicating that this subspecies detected in human fecal samples may not colonize in the human gut but was supplied as probiotics. However, the volunteer 2 had very similar strains that were sampled at a 10-year interval. This result suggested possibilities that the dietary habits of this volunteer, such as yoghurt intake, had not changed during the 10 years intervention, or that B. animalis subsp. lactis strains had colonized in his gut.
To investigate whether strains isolated from volunteer 2 were due to his dietary habits, we compared the number of SNPs between the nucleotide sequences of 11 reference genomes obtained by downloading from NCBI FTP site and that of two genomes isolated from volunteer 2. As the result, the average number of the SNPs between two strains was 281 (the minimum number of SNPs was 22 and the maximum number of that was 814: Figure 2). In Japan, two dairy products containing B. animalis subsp. lactis are sold: DANONE BIO® (Danone Japan Co., Ltd., Tokyo, Japan), which contains B. animalis subsp. lactis CNCM I-2494 (https://www.danone.co.jp/bio/about/powerofbe80/be80/; accessed 24 September 2019) and BifiX® (EZAKI GLICO Co., Ltd., Osaka, Japan), which contains B. animalis subsp. lactis GCL2505 (https://www.glico.co.jp/laboratory/bifix/02.html; accessed 24 September 2019). The number of SNPs between CNCM I-2494 and strains isolated from volunteer 2 (2-Y0; 312, 2-Y10; 323) were more than the average of SNPs (281), which implies that strains isolated from volunteer 2 were not supplied by DANONE BIO® (Figure 2). The genome of B. animalis subsp. lactis GCL2505 is not registered to NCBI FTP site. Probiotic bacterium supplied by commercial product is probably an identical strain. SNPs between strains isolated from volunteer 2 was 27, which was more than SNPs between Bl-07 and Bi-04. These two strains were differently isolated, and phylogenetically close relationship is consistent with a previous study [26]. The result supports that strains isolated from volunteer 2 were not supplied by dietary habits. Detailed survey on dietary habits and a larger number of volunteers are required to conclude whether a strain of B. animalis subsp. lactis colonize in the human gut.
Heatmap showing the number of single nucleotide polymorphisms (SNPs) in nucleotide sequences of genomes isolated from volunteer 2 and 11 publicly available B. animalis subsp. lactis strains. The number in matrix represents the number of putative SNPs between two strains calculated by Mauve. The color represents abundance of SNPs.
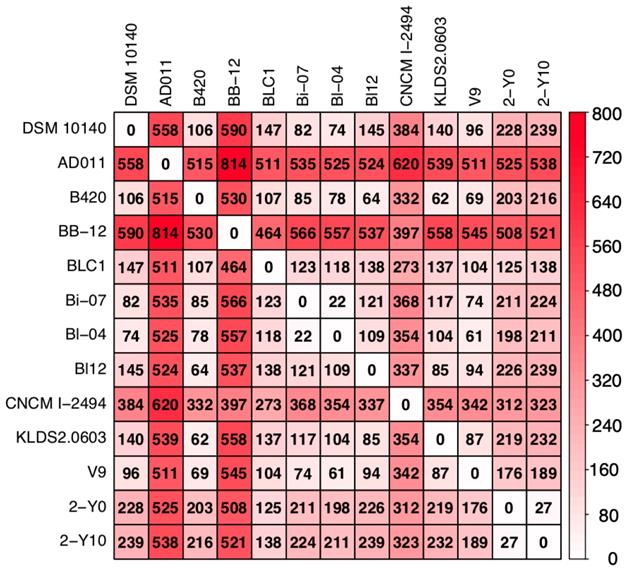
Previous studies related to the B. animalis in the human gut were mainly based on the 16S rRNA amplicon sequence [27-29]. This paper is the first report of draft genome sequences of B. animalis that includes multiple samples isolated from a single Japanese individual, although the study involved only two volunteers. These results provide insight into the in vivo colonization of Bifidobacterium and personalized probiotic supplementation.
Nucleotide sequence accession number
The draft genome sequence of the three strains isolated from the volunteers (1-Y0, 2-Y0, and 2-Y10) has been deposited at GenBank/EMBL/DDBJ under BioProject number PRJDB8215, BioSample number SAMD00168440, SAMD00168441, and SAMD00168442 respectively, and accession number BJKG00000000, BJKH00000000, and BJKI00000000 respectively (accession range: BJKG01000001-BJKG01000015, BJKH01000001-BJKH01000015, and BJKI01000001-BJKI01000015). The version described in this paper is the first version (BJKG01000000, BJKH01000000, and BJKI01000000). The raw reads have been deposited in the DDBJ Sequence Read Archive (DRA) under Submission DRA008418.
Abbreviations
SCFA: short-chain fatty acid; PBS: phosphate-buffered saline; TOS: transgalactosylated oligosaccharide; MRS: De Man, Rogosa and Sharpe; SNPs: single nucleotide polymorphisms.
Acknowledgements
We thank Ms. Yuka Ohara, Mrs. Mitsuko Komatsu, and Noriko Kagata for technical support; Drs. Yasuyuki Ohkawa, Tetsuya Hayashi, Yasuhiro Gotoh, and Keiji Nakamura for genome sequencing; Dr. Yoshitoshi Ogura for data analysis; Drs. Jiayue Yang, Shinnosuke Murakami, Naoki Tanigawa, Yongshou Yang, Chiharu Ishii, and Ms. Nao Takeuchi for critical discussions. This work was partly performed in the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University. This work was supported in part by JSPS KAKENHI (grant no. 16H04901, 17H05654, and 18H04805 to S.F.), JST PRESTO (grant no. JPMJPR1537 to S.F.), JST ERATO (grant no. JPMJER1902 to S.F.), AMED-CREST (grant no. JP19gm1010009 to S.F.), the Takeda Science Foundation (to S.F.), the Food Science Institute Foundation (to S.F.), the Program for the Advancement of Research in Core Projects under Keio University's Longevity Initiative (to S.F.), the Yamagata Prefectural Government and the city of Tsuruoka (to M.T.), and Taikichiro Mori Memorial Research Fund (to T.T.). Sequence analysis in this study was supported by JSPS KAKENHI (grant no. 16H06279 to PAGS).
Ethics Committee Approval and Patient Consent
This study was approved by the Ethics Committee of Keio University Shonan Fujisawa Campus and Kensyou-kai Incorporated Medical Institution under the approval number 195 and 20170915-4, respectively. Both subjects were informed of the purpose of this study, and written consent was obtained from both subjects.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLOS Biol. 2016;14:e1002533
2. Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K. et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469:543-7
3. Bindels LB, Porporato P, Dewulf EM, Verrax J, Neyrinck AM, Martin JC. et al. Gut microbiota-derived propionate reduces cancer cell proliferation in the liver. Br J Cancer. 2012;107:1337-44
4. Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B. et al. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11:506-14
5. Biavati B, Mattarelli P. The prokaryotes, vol 3. New York, NY: Springer New York. 2006
6. De Vuyst L, Leroy F. Cross-feeding between bifidobacteria and butyrate-producing colon bacteria explains bifdobacterial competitiveness, butyrate production, and gas production. Int J Food Microbiol. 2011;149:73-80
7. Maldonado-Gómez MX, Martínez I, Bottacini F, O'Callaghan A, Ventura M, van Sinderen D. et al. Stable Engraftment of Bifidobacterium longum AH1206 in the Human Gut Depends on Individualized Features of the Resident Microbiome. Cell Host Microbe. 2016;20:515-26
8. Parte AC. LPSN - List of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2014;42:D613-D616
9. Liu Q, Jing W, Wang W. Bifidobacterium lactis Ameliorates the Risk of Food Allergy in Chinese Children by Affecting Relative Percentage of Treg and Th17 Cells. Can J Infect Dis Med Microbiol. 2018;2018:1-12
10. Wildt S, Nordgaard I, Hansen U, Brockmann E, Rumessen JJ. A randomised double-blind placebo-controlled trial with Lactobacillus acidophilus La-5 and Bifidobacterium animalis subsp. lactis BB-12 for maintenance of remission in ulcerative colitis. J Crohn's Colitis. 2011;5:115-21
11. Ushida K, Uwatoko Y, Adachi Y, Soumah AG, Matsuzawa T. Isolation of bifidobacteria from feces of chimpanzees in the wild. J Gen Appl Microbiol. 2010;56:57-60
12. Watanabe K, Fujimoto J, Matsuki T, Matsumoto K, Kado Y, Takada T. et al. Quantitative PCR with 16S rRNA-Gene-Targeted Species-Specific Primers for Analysis of Human Intestinal Bifidobacteria. Appl Environ Microbiol. 2004;70:167-73
13. Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014;24:1384-95
14. Simão FA, Waterhouse RM, Ioannidis P, Kriventseva E V, Zdobnov EM. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210-2
15. Nishimura O, Hara Y, Kuraku S. GVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics. 2017;33:3635-7
16. Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072-5
17. Tanizawa Y, Fujisawa T, Nakamura Y. DFAST: A flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics. 2018;34:1037-9
18. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403-10
19. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K. et al. BLAST+: Architecture and applications. BMC Bioinformatics. 2009;10:421
20. Wong VK, Reuter S, Hunt M, Holden MTG, Fookes M, Page AJ. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691-3
21. Nakamura T, Yamada KD, Tomii K, Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics. 2018;34:2490-2
22. Price MN, Dehal PS, Arkin AP. FastTree 2 - Approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490
23. Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394-403
24. Dudley EG, Broadbent JR, Rendulic S, Schuster S, Roberts RF, Loquasto JR. et al. Comparison of the Complete Genome Sequences of Bifidobacterium animalis subsp. lactis DSM 10140 and Bl-04. J Bacteriol. 2009;191:4144-51
25. Turroni F, Foroni E, Pizzetti P, Giubellini V, Ribbera A, Merusi P. et al. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Appl Environ Microbiol. 2009;75:1534-45
26. Milani C, Duranti S, Lugli GA, Bottacini F, Strati F, Arioli S. et al. Comparative genomics of Bifidobacterium animalis subsp. lactis reveals a strict monophyletic bifidobacterial taxon. Appl Environ Microbiol. 2013;79:4304-15
27. Kim SW, Suda W, Kim S, Oshima K, Fukuda S, Ohno H. et al. Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res. 2013;20:241-53
28. Malinen E, Matto J, Salmitie M, Alander M, Saarela M, Palva A. PCR-ELISAII: Analysis of Bifidobacterium populations in human faecal samples from a consumption trial with Bifidobacterium lactis Bb-12 and a galacto-oligosaccharide preparation. Syst Appl Microbiol. 2006;25:249-58
29. Bogovič Matijašić B, Obermajer T, Lipoglavšek L, Sernel T, Locatelli I, Kos M. et al. Effects of synbiotic fermented milk containing Lactobacillus acidophilus La-5 and Bifidobacterium animalis ssp. lactis BB-12 on the fecal microbiota of adults with irritable bowel syndrome: A randomized double-blind, placebo-controlled trial. J Dairy Sci. 2016;99:5008-21
Author contact
![]() Corresponding author: Shinji Fukuda, Institute for Advanced Biosciences, Keio University, 246-2 Mizukami, Kakuganji, Tsuruoka, Yamagata 997-0052, JAPAN. Email: sfukudakeio.ac.jp; TEL: +81-235-29-0528; FAX: +81-235-29-0574
Corresponding author: Shinji Fukuda, Institute for Advanced Biosciences, Keio University, 246-2 Mizukami, Kakuganji, Tsuruoka, Yamagata 997-0052, JAPAN. Email: sfukudakeio.ac.jp; TEL: +81-235-29-0528; FAX: +81-235-29-0574