ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2022; 10:45-48. doi:10.7150/jgen.69822 This volume Cite
Research Paper
Complete Genome Sequence of Four Strains of Leptospira borgpetersenii serovar Hardjo isolated from Cattle in the Central United States
Ellie J. Putz, Darrell O. Bayles, David P. Alt, Jarlath E. Nally ![]()
Infectious Bacterial Disease Research Unit, USDA Agriculture Research Service, National Animal Disease Center, Ames, IA, USA.
Received 2021-12-7; Accepted 2022-1-24; Published 2022-2-14
Abstract

Pathogenic species of Leptospira cause leptospirosis, a global zoonotic disease affecting humans and all major livestock species. Cattle act as a reservoir host for L. borgpetersenii serovar Hardjo which colonize the kidneys and reproductive tract from which they are excreted and transmitted to other cattle via urine, semen or uterine discharges. Bovine leptospirosis results in reproductive failure, abortion, stillbirth and loss of milk production, and is an occupational risk for those working with infected animals. A recent study determined that 7.2% of cattle from an abattoir in the central United States were actively shedding pathogenic Leptospira. Here, we report and compare the complete genome sequence of four recent isolates of L. borgpetersenii serovar Hardjo designated strain TC112, TC147, TC129, and TC273.
Keywords: Leptospira borgpetersenii, leptospirosis, bovine, PanOCT/PanACEA
Introduction
Leptospirosis is a global zoonotic disease caused by bacteria of the genus Leptospira. Over one million people suffer acute leptospirosis each year, and almost 60,000 people die [1]. Leptospirosis is also a significant cause of morbidity and mortality in agricultural livestock animals since infected animals can present with acute disease as well as abortion, reproductive failure and weak offspring [2]. Transmission of leptospires is maintained by reservoir hosts of infection, which shed leptospires exposing other hosts directly via urine, semen, and uterine discharges, or indirectly via contamination of moist environments [3]. Leptospira are classified into 38 pathogenic species (clade P1 and P2) and comprise hundreds of serovars in multiple serogroups [4, 5]. Multiple serovars of L. interrogans are the leading global cause of leptospirosis in humans. However, L. borgpetersenii serovar Hardjo is the leading global cause of bovine leptospirosis, with cattle serving as the primary reservoir host [2]. The genome of L. borgpetersenii is ~700kb smaller than that of L. interrogans and is hypothesized to be undergoing genome reduction [6].
Bovine leptospirosis is endemic in the United States [7-9]. During a recent survey in cattle from an abattoir in the Midwestern United States, it was found that 7.2% of animals were actively shedding L. borgpetersenii serovar Hardjo, by fluorescent antibody testing, PCR and culture of urine [8, 10]. Four isolates (designated strains TC112, TC129, TC147, and TC273) cultured from bovine urine during that study have now been fully sequenced and are compared herein. All genome sequence information is available on GenBank (https://www.ncbi.nlm.nih.gov/genbank/), BioProject PRJNA759631. Accession numbers for chromosome 1 and chromosome 2 for each of the four strains, as well as genome annotation features, are provided in Table 1.
Genome annotation of L. borgpetersenii serovar Hardjo strains
| TC112 | TC129 | TC147 | TC273 | HB203* | |
|---|---|---|---|---|---|
| Accession Number (Chromosome 1 & 2) | CP084036 CP084037 | CP084038 CP084039 | CP084040 CP084041 | CP084042 CP084043 | CP021412 CP021413 |
| Chromosome 1 (bp) | 3,585,599 | 3,585,597 | 3,585,599 | 3,585,577 | 3,589,981 |
| Chromosome 2 (bp) | 317,347 | 317,349 | 317,349 | 317,347 | 317,347 |
| G+C % | 40 | 40 | 40 | 40 | 40 |
| Genes (total) | 3,389 | 3,403 | 3,403 | 3,403 | 3,539 |
| CDSs (total) | 3,345 | 3,359 | 3,359 | 3,359 | 3,495 |
| Genes (coding) | 2,945 | 2,962 | 2,962 | 2,958 | 3,079 |
| CDSs (with protein) | 2,945 | 2,962 | 2,962 | 2,958 | 3,079 |
| Genes (RNA) | 44 | 44 | 44 | 44 | 44 |
| rRNAs (5S, 16S, 23S) | 1, 2, 2 | 1, 2, 2 | 1, 2, 2 | 1, 2, 2 | 1, 2, 2 |
| tRNAs | 37 | 37 | 37 | 37 | 37 |
| ncRNAs | 2 | 2 | 2 | 2 | 2 |
| Pseudo Genes (total) | 400 | 397 | 397 | 401 | 416 |
*Strain of L. borgpetersenii serovar Hardjo originally isolated from bovine over 30 years ago [7].
All strains were cultured at 29 °C until they reached the mid-late log growth phase in HAN media [11]. Genome sequencing on the Illumina platform was performed as previously described [8]. For genome sequencing on the Nanopore platform, ten ml of culture was pelleted by centrifugation at 7,000 × g, for 20 min at 4 °C. The supernatant was removed, and the pellet resuspended in 1 ml Phosphate-buffered saline (PBS), placed in a 1.5 ml Protein LoBind tube (Eppendorf, Hamburg, Germany) and pelleted by centrifugation at 12,000 × g, for 10 min at 4 °C. The supernatant was removed, and the pellet resuspended in 20 µl PBS. DNA was purified from each of the pellets with the Circulomics Nanobind CBB Big DNA Kit (Circulomics, Baltimore, MD, USA) using the Gram-Negative Bacteria High Molecular Weight DNA Extraction protocol. Purified DNA was quantified using the Thermo Fisher Qubit™ dsDNA BR Assay Kit (Thermo Fisher Scientific, Waltham, MA) and evaluated for quality and purity using a NanoPhotometer Pearl (Implen, Inc. Westlake Village, CA). Libraries for strains TC129 and TC273 were prepared using the ligation sequencing kit, (SQK-LSK-109), multiplexed using native barcoding expansion kit (EXP-NBD104) and sequenced using flow cell type R9.4.1 on a GridION 5X instrument (Oxford Nanopore, Oxford Science Park, UK). Libraries for strains TC147 and TC273 were prepared using the rapid barcoding kit (SQK-RBK004) and sequenced using the flongle flow cell type FLO-FLG001 on an MK1c instrument (Oxford Nanopore, Oxford Science Park, UK). Genome assembly and error correction was accomplished with Unicycler v. 0.4.4 (TC129 and TC273) and v. 0.4.7 (TC112 and TC147) [12]. Oxford Nanopore and unpaired Illumina HiSeq reads were used with the Unicycler default settings except that a Leptospira ParA protein sequence was added to the start genes database to ensure that the small chromosome would be rotated to start at the parA gene. The genome annotation was completed by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) [13].
Comparative analysis of the complete genomes for each of the four recent L. borgpetersenii serovar Hardjo TC strains was performed against the complete genome of another bovine isolate of L. borgpetersenii serovar Hardjo; strain Hb15B203 (HB203) which was originally isolated from a 10-year old dairy cow from Kansas over 30 years ago [7]. Since its isolation, strain HB203 has been utilized extensively as a challenge isolate in animal work for the Infectious Bacterial Disease Research unit (ARS, USDA) and its complete genome is also available on GenBank (BioProject: PRJNA384237) [14-18].
Comparison of all genomes using PanOCT (pan-genome ortholog clustering tool) and PanACEA (Pan-genome Atlas with Chromosome Explorer and Analyzer) identified differences in gene coding regions of the complete genome of strain HB203 and the more recently isolated strains TC112, TC129, TC147, and TC273 [19, 20], Figure 1. In total, strain HB203 contained 206 genes not identified in any of the more recent TC strains. Conversely, 74 genes were identified in TC112, 90 genes in TC129, 88 genes in TC147 and 86 genes in TC273 that were not identified in strain HB203. A complete list of matching genes among all strains is provided in Supplemental Table 1. Of note, some variances in the PGAP pipeline's annotation of open reading frames (ORFs) for each of the TC strains were identified; For example, TC129 contained 14 small (averaging 57 amino acid) CDS which were not annotated in TC112 yet there are corresponding potential open reading frames of exactly the same size and with the same predicted amino acid for each one. After removal of these small ab initio gene calls from analysis, as listed Supplementary Table 2, and removal of some additional annotation calls for which homologous DNA was found in all TC strains (highlighted in yellow in supplementary Table 1), there were 71 genes identified in all four TC strains that had no homologous gene in HB203.
Comparison of the genomes of L. borgpetersenii serovar Hardjo strains TC112, TC129, TC147, TC273 and HB203 with PanACEA. Shown are (A) chromosome 1 and (B) chromosome 2. Using HB203 as the reference, the light gray color depicts core conserved regions and the dark gray lines within outermost circle depict variable regions between strains.
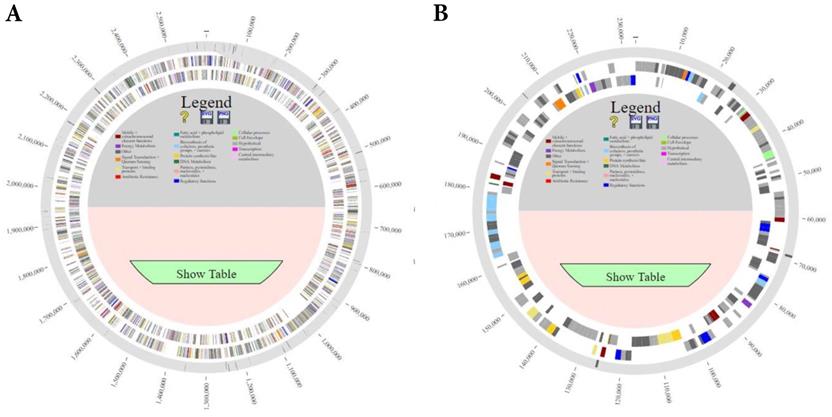
Globally, bovines are a reservoir host for L. borgpetersenii serovar Hardjo which are highly fastidious [2]; a limited number of closed genomes have been sequenced but are poorly annotated. Of the 206 genes identified by PanOCT/PanACEA as unique to strain HB203, 134 (>65%) are annotated as encoding hypothetical proteins. Similarly, of the 71 genes unique to the TC strains, 32 (>45%) encode hypothetical proteins. As additional genomes are sequenced, comparative genome analyses with other strains obtained from different parts of the world, such as strain L550 in Australia [6], and separated by time, can provide insights into how this species and serovar continues to evolve and persistently infect cattle despite the availability of bacterin vaccines.
Genome comparisons using the single nucleotide polymorphisms (SNPs) calling capabilities of Mauve software [21] (Mauve development snapshot 2015-02-13; http://darlinglab.org/mauve/mauve.html) identified just 226 SNPs between the four novel TC strains and HB203. The SNP differences and their position in the respective genome are provided in Supplemental Table 3.
Collectively, these data demonstrate that the genomes of L. borgpetersenii serovar Hardjo strains TC112, TC129, TC147, and TC273 are highly similar to each other and to strain HB203 even though HB203 was isolated approximately 30 years earlier. Bovine leptospirosis due to L. borgpetersenii serovar Hardjo is a global disease. This work provides four additional complete genomes of recent bovine isolates of L. borgpetersenii serovar Hardjo obtained from U.S. cattle which will facilitate continued comparative genome analysis with other serovar Hardjo isolates, separated over time and geography, to identify conserved genome features, understand evolutionary traits that predispose serovar Hardjo to persistent carriage in cattle and zoonotic transmission, and gain additional insights into pathogenic mechanisms of infection.
Supplementary Material
Supplementary tables.
Acknowledgements
The authors thank the Iowa State University DNA facility for sample preparation and library creation. USDA is an equal opportunity provider and employer. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Data availability
Annotated assemblies are available in GenBank, BioProject PRJNA759631: Accession numbers for each strain are as follows: TC112 (chromosome 1 Accession: CP084036, chromosome 2 Accession: CP084037), TC129 (chromosome 1 Accession: CP084038, chromosome 2 Accession: CP084039), TC147 (chromosome 1 Accession: CP084040, chromosome 2 Accession: CP084041), and TC273 (chromosome 1 Accession: CP084042, chromosome 2 Accession: CP084043).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Costa F, Hagan JE, Calcagno J, Kane M, Torgerson P, Martinez-Silveira MS. et al. Global morbidity and mortality of leptospirosis: a systematic review. PLoS Neglected Tropical Diseases. 2015;9:e0003898
2. Ellis WA. Animal leptospirosis. Current Topics in Microbiology and Immunology. 2015;387:99-137
3. Putz EJ, Nally JE. Investigating the immunological and biological equilibrium of reservoir hosts and pathogenic Leptospira: balancing the solution to an acute problem? Frontiers in Microbiology. 2020;11:2005
4. Vincent AT, Schiettekatte O, Goarant C, Neela VK, Bernet E, Thibeaux R. et al. Revisiting the taxonomy and evolution of pathogenicity of the genus Leptospira through the prism of genomics. PLoS Neglected Tropical Diseases. 2019;13:e0007270
5. Faine S, Adler B, Bolin C, Perolat P. Leptospira and Leptospirosis. 2 ed. Melbourne, Australia: MediSci. 1999
6. Bulach DM, Zuerner RL, Wilson P, Seemann T, McGrath A, Cullen PA. et al. Genome reduction in Leptospira borgpetersenii reflects limited transmission potential. Proc Natl Acad Sci U S A. 2006;103:14560-5
7. Miller D, Wilson M, Beran G. Survey to estimate prevalence of Leptospira interrogans infection in mature cattle in the United States. American Journal of Veterinary Research. 1991;52:1761-5
8. Nally JE, Hornsby RL, Alt DP, Bayles D, Wilson-Welder JH, Palmquist DE. et al. Isolation and characterization of pathogenic leptospires associated with cattle. Veterinary Microbiology. 2018;218:25-30
9. Talpada MD, Garvey N, Sprowls R, Eugster AK, Vinetz JM. Prevalence of leptospiral infection in Texas cattle: implications for transmission to humans. Vector-Borne and Zoonotic Diseases. 2003;3:141-7
10. Nally JE, Ahmed AA, Putz EJ, Palmquist DE, Goris MG. Comparison of real-time PCR, bacteriologic culture and fluorescent antibody test for the detection of Leptospira borgpetersenii in Urine of naturally infected cattle. Veterinary Sciences. 2020;7:66
11. Hornsby RL, Alt DP, Nally JE. Isolation and propagation of leptospires at 37 degrees C directly from the mammalian host. Sci Rep. 2020;10:9620
12. Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Computational Biology. 2017;13:e1005595
13. Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L. et al. NCBI prokaryotic genome annotation pipeline. Nucleic acids research. 2016;44:6614-24
14. Zuerner RL, Alt DP, Palmer MV, Thacker TC, Olsen SC. A Leptospira borgpetersenii serovar Hardjo vaccine induces a Th1 response, activates NK cells, and reduces renal colonization. Clin Vaccine Immunol. 2011;18:684-91
15. Zuerner R, Alt D, Palmer M. Development of chronic and acute golden Syrian hamster infection models with Leptospira borgpetersenii serovar Hardjo. Veterinary Pathology. 2012;49:403-11
16. Bolin C, Thiermann A, Handsaker A, Foley J. Effect of vaccination with a pentavalent leptospiral vaccine on Leptospira interrogans serovar hardjo type hardjo-bovis infection of pregnant cattle. American Journal of Veterinary Research. 1989;50:161-5
17. Bolin CA, Alt DP. Use of a monovalent leptospiral vaccine to prevent renal colonization and urinary shedding in cattle exposed to Leptospira borgpetersenii serovar hardjo. American Journal of Veterinary Research. 2001;62:995-1000
18. Bolin C, Zuerner R, Trueba G. Effect of vaccination with a pentavalent leptospiral vaccine containing Leptospira interrogans serovar hardjo type hardjo-bovis on type hardjo-bovis infection of cattle. American Journal of Veterinary Research. 1989;50:2004-8
19. Inman JM, Sutton GG, Beck E, Brinkac LM, Clarke TH, Fouts DE. Large-scale comparative analysis of microbial pan-genomes using PanOCT. Bioinformatics. 2019;35:1049-50
20. Clarke TH, Brinkac LM, Inman JM, Sutton G, Fouts DE. PanACEA: a bioinformatics tool for the exploration and visualization of bacterial pan-chromosomes. BMC Bioinformatics. 2018;19:246
21. Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Research. 2004;14:1394-403
Author contact
![]() Corresponding author: E-mail: jarlath.nallygov.
Corresponding author: E-mail: jarlath.nallygov.