ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2022; 10:69-77. doi:10.7150/jgen.76121 This volume Cite
Research Paper
Draft Genomes Sequences of 11 Geodermatophilaceae Strains Isolated from Building Stones from New England and Indian Stone Ruins found at historic sites in Tamil Nadu, India
Nathaniel J. Ennis1,5, Dhanasekaran Dharumadurai1,2, Joseph L. Sevigny1,3, Ryan Wilmot1, Sulaiman M. Alnaimat1,6, Julia G. Bryce4, W. Kelley Thomas1,3, Louis S. Tisa1 ![]()
1. Department of Molecular, Cellular, and Biomedical Sciences, University of New Hampshire, Durham, New Hampshire, USA.
2. Departments of Microbiology, Bharathidasan University, Tiruchirappalli, Tamil Nadu, India.
3. Hubbard Center for Genome Studies, University of New Hampshire, Durham, New Hampshire, USA.
4. Department of Earth Sciences, University of New Hampshire, Durham, NH, USA.
5. Present address: Seres Therapeutics, Cambridge, MA, USA.
6. Present address: Department of Medical Analysis, Al-Hussein Bin Talal University, Ma'an, Jordan.
Received 2022-6-14; Accepted 2022-8-26; Published 2022-9-21
Abstract

Metagenomic analysis of stone microbiome from samples collected in New England, USA and Tamil Nadu, India identified numerous Actinobacteria including Geodermatphilaceae. A culture-dependent approach was performed as a companion study with this culture-independent metagenomic analysis of these stone samples and resulted in the isolation of eleven Geodermatphilaceae strains (2 Geodermatophilus and 9 Blastococcus strains). The genomes of the 11 Geodermatphilaceae strains were sequenced and analyzed. The genomes for the two Geodermatophilus isolates, DF1-2 and TF2-6, were 4.45 and 4.75 Mb, respectively, while the Blastococcus genomes ranged in size from 3.98 to 5.48 Mb. Phylogenetic analysis, digital DNA:DNA hybridization (dDDH), and comparisons of the average nucleotide identities (ANI) suggest the isolates represent novel Geodermatophilus and Blastococcus species. Functional analysis of the Geodermatphilaceae genomes provides insight on the stone microbiome niche.
Keywords: Genomes, Stones, Ruins, Climate, Geochemistry, Geodermatophilaceae, Actinobacteria.
Introduction
Stone surfaces provide a harsh environment with limited nutrient and water availability, exposure to lethal UV irradiation, potential contact with toxic metals and metalloids, and cycles in temperature variation [1-4]. Despite these seemly inhospitable conditions, stone surfaces can support microbial life and well-defined communities. Because of their hyphal nature, Actinobacteria have been considered a primary colonizer of rock that then helps promote the growth of successive microbial colonizers. Members of the family Geodermatophilaceae have also been consistently isolated from stone surfaces and interiors [5].
We have been investigating the stone microbiome across a variety of lithologies three sites (North Africa, Southern Tamil Nadu, India and New England, USA) using culture-independent metagenomic approaches [3, 6, 7]. To supplement this metagenomic approach, a culture-dependent approach was taken to isolate Actinobacteria from two of these sites (Southern Tamil Nadu, India and New England, USA). This study focuses on the genomes of Blastococcus and Geodermatophilus, two genera of the family Geodermatophilaceae, of bacterial strains that were isolated from samples obtained at these sites.
Material and Methods
Stone samples
Stone samples were obtained from historic sites in Tamil Nadu, India and at three different colonial sites in New England [6, 7]. These stone samples were used in culture-independent studies to determine the stone microbiome structure [6, 7]. These samples were also used to obtain bacterial isolates for culture-dependent studies.
Isolation of Bacteria Associated with Stone Surfaces
Stone samples were crushed aseptically with a surface-sterilized rock hammer in a Biosafety hood. Crushed rock samples were reduced to a powder by grinding with a sterile mortar and pestle. The pulverized stone samples were used to isolate stone-dwelling bacteria. Table 1 shows the stone samples and other pertinent information on the 11 Geodermatophilaceae isolates used in this study. For this approach, pulverized stone (0.5g) was suspended in 5 mL of sterile phosphate-buffered saline (PBS) solution [8] and mixed thoroughly on a vortex mixer for 1 min. Stone suspensions were serially diluted in PBS from 10-1 to 10-6 dilutions. For each stone sample, 100 uL of the 10-4, 10-5, and 10-6 dilutions were spread plated onto the following media types: Czapek supplemented with yeast extract (DSMZ medium 130; [9]), Luedemann agar (DSMZ medium 877; [10]), R2A agar (DSMZ medium 830; [11]), and Starch Casein agar [12]. Cycloheximide (50 ug/mL final concentration) was added to the growth media to inhibit fungal growth. These growth media were chosen to select for Actinobacteria or other slow-growing bacteria [9, 11]. The plates were sealed with parafilm to retain moisture and were incubated at 28oC for two months before attempting to isolate individual colonies. Colonies were chosen for isolation based primarily on pigmentation indicating UV tolerance, but also based on distinct colony morphology and slow growth rate (one week or more of incubation needed for colony growth). Individual colonies were purified on the same medium that isolation was accomplished. All purified isolates were grown for three to five days in their appropriate medium and prepared for long-term storage at -80oC by mixing the culture with an equal volume of 60% glycerol. Among the two sampling regions, a total of 85 bacterial isolates were purified, identified, and stored.
Geodermatophilaceae isolates used in this study and information on the stone sample.
| Isolate | Medium1 | Specific site of collection | Location | Coordinate (DMS) | Stone type | Climate | Stone Condition | Approximate Stone Age (Years) |
|---|---|---|---|---|---|---|---|---|
| TF02-8 | Czpek | Outside rock damage area | Fort Tiruchirappalli, Tamil Nadu, India | 10o49'40” N 78o41'49” E | Granite | Tropical Wet and Dry | Built | 1,000-1,500 |
| TF02-6 | Czpek | Outside rock damage area | Fort Tiruchirappalli, Tamil Nadu, India | 10o49'40” N 78o41'49” E | Granite | Tropical Wet and Dry | Built | 1,000-1,500 |
| TF02-09 | Czpek | Outside rock damage area | Fort Tiruchirappalli, Tamil Nadu, India | 10o49'40” N 78o41'49” E | Granite | Tropical Wet and Dry | Built | 1,000-1,500 |
| TF02A-26 | Czpek | Temple wall outside | Fort Tiruchirappalli, Tamil Nadu, India | 10o49'40” N 78o41'49” E | Granite | Tropical Wet and Dry | Built | 1,000-1,500 |
| TF02A-30 | Czpek | Temple wall outside | Fort Tiruchirappalli, Tamil Nadu, India | 10o49'40” N 78o41'49” E | Granite | Tropical Wet and Dry | Built | 1,000-1,500 |
| TF02A-35 | Czpek | Temple wall outside | Fort Tiruchirappalli, Tamil Nadu, India | 10o49'40” N 78o41'49” E | Granite | Tropical Wet and Dry | Built | 1,000-1,500 |
| TBT05-19 | Czpek | Temple wall outside damage area | Thanjavur Big Temple, Tamil Nadu, India | 10o46'58” N 79o7'54” E | Granite | Tropical Wet and Dry | Built | 1,000-1,500 |
| DF01-2 | Czapek | Temple wall outside | Fort Dindigul, Tamil Nadu, India | 10o21'39” N 77o57'42” E | Granite | Tropical Wet and Dry | Built | 250-500 |
| CT_GayMR16 | R2A | Mill site foundation | Gay City State Park Hebron, CT, USA | 41o43'34” N 72o26'24” W | Granite | Humid Continental | Built | 150-200 |
| CT_GayMR19 | LDM | Mill site foundation | Gay City State Park Hebron, CT, USA | 41o43'34” N 72o26'24” W | Granite | Humid Continental | Built | 150-200 |
| CT_GayMR20 | LDM | Mill site foundation | Gay City State Park Hebron, CT, USA | 41o43'34” N 72o26'24” W | Granite | Humid Continental | Built | 150-200 |
Extraction of Genomic DNA from Bacterial Isolates
Isolates were grown for three to five days in Czapek broth supplemented with yeast extract. Genomic DNA (gDNA) of the bacterial stone isolates was extracted by the cetyl trimethylammonium bromide (CTAB) method [13]. The extracted gDNA was suspended in Tris-EDTA (TE) buffer and treated with RNase to remove RNA. The extracted DNA was quantified using a Nanodrop 2000c (Thermo Fisher Scientific, Waltham, MA).
Amplification of Bacterial Isolate 16S rRNA Genes
To identify the isolated stone-dwelling bacteria, the 16S rRNA gene of each isolate was amplified through PCR using the extracted gDNA of each isolate. The gDNA was combined with OneTaq Hot Start Polymerase (New England Biolabs, Ipswich, MA) and primers A 7-26f (5'-CCG-TCG-ACG-AGC-TCA-GAG-TTT-GAT-CCT-GGC-TCA-3') and B 1523-1504r (5'-CCC-GGG-TAC-CAA-GCT-TAA-GGA-GGT-GAT-CCA-GCC-GCA-3'), as described previously [14]. The conditions for thermal cycling were as follows: an initial denaturation step at 95oC for 5 min was followed by 35 cycles of denaturation at 95oC for 30 s, primer annealing at 55oC for 30 s, and extension at 68oC for 2 min, with the final cycle followed by a 10 min extension at 68oC. The amplified PCR products were purified using the QiaQuick PCR Purification Kit following the manufacturer's protocol (Qiagen, Hilden, Germany). The presence and approximate size of the 16S gene was verified through gel electrophoresis. Amplified PCR products were quantified using the Qubit Fluorometric Quantitation system (Thermo Fisher Scientific, Waltham, MA).
Sanger Sequencing and Identification of Isolate 16S rRNA Genes
To obtain approximate identities of all stone-dwelling bacterial isolates, partial sequences corresponding to the mid-region of isolate 16S genes were obtained by Sanger Sequencing [15] through Genewiz according to the service guidelines (Genewiz Inc., South Plainfield, NJ) and using primer 907r (5'-CCG-TCA-ATT-CCT-TTR-AGT-TT-3'), as described previously [16]. Partial sequences were aligned with the 16S ribosomal RNA sequence (Bacteria and Archaea) database using the Basic Local Alignment Search Tool (BLAST), through blastn Version 2.7.1 (NCBI, Bethesda, MD). Isolates were identified as the BLAST result with the highest alignment score.
The full 16S rRNA gene of isolates that were identified as being closely related to members of the Actinobacteria family Geodermatophilaceae was generated by Sanger sequencing as described above and by using additional sequencing primers to ensure coverage of the full 16S rRNA gene. The sequencing primers used were: A 7-26f, B 1523-1504r, C 704-685r (5'-TCT-GCG-CAT-TTC-ACC-GCT-AC-3') and D 1115-1100r (5'-AGG-GTT-GCG-CTC-GTT-G-3'), as described previously [14]. The sequences for each of Geodermatophilaceae isolate were aligned to build a final consensus sequence of the full 16S rRNA gene using Serial Cloner Version 2.6.1 (Serial Basics, 2013). Full 16S rRNA gene sequences were aligned using BLAST as described above, and isolates were more accurately identified as the BLAST result with the highest alignment score.
Sequences of the full 16S rRNA genes of each Geodermatophilaceae isolate were submitted to GenBank [17] to add to the repository of publicly available DNA sequences and for future potential publication of novel isolates. GenBank accession numbers are MK239636-MK239646.
Whole Genome Shotgun Sequencing of Geodermatophilaceae Isolates
To fully identify and explore the functional capacity of potentially novel Geodermatophilaceae isolates, whole genome shotgun sequencing was performed on the gDNA of the stone isolates identified as members of Geodermatophilaceae according to the 16S rRNA sequencing described above. Sequencing libraries for the eleven Geodermatophilaceae isolates were prepared using the Illumina Nextera Library Preparation protocol according to the manufacturer's instructions (Illumina Inc., San Diego, CA). Sequencing was completed on an Illumina HISeq 2500 HiSeq2500 platform (Illumina Inc., San Diego, CA) to produce 250 bp paired-end reads at the Hubbard Center for Genome Studies (UNH, Durham, NH). Raw sequencing data was demultiplexed using bcl2convert.
Quality Filtering of Whole Genome Shotgun Sequencing Reads
Sequence data were trimmed using Trimmonatic version 0.36 [18]. Truseq adapters were trimmed with an allowance of two mismatches. Leading and trailing bases below quality of three were trimmed. The read was then scanned with a sliding window of 4 bps and trimmed if the average quality dropped below 30. Finally, reads were dropped if the length was less than 36 bps. Trimmed sequencing reads were assembled using SPAdes version 3.13 [19] with default settings. The assembled genomes were annotated via the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) [20]. The assembly metrics and annotation features are given in Table 2. The identities of the strains were determined by a whole genome-based taxonomic analysis via the Type (Strain) Genome Server (TYGS) platform [21] (https://tygs.dsmz.de) including digital DNA:DNA hybridization (dDDH) values [22]. Average nucleotide identity (ANI) analysis of these genomes was performed on the JSpeciesWS server (https://jspecies.ribohost.com/jspeciesws/) [23].
Genome Statistics.
| Bacterial species | Isolate | Genebank accession no. | Numbers of reads | No. of contigs | Avg coverage (X) | Genome assembly size (bp) | N50 contig size (kb) | No. of CDSs | G + C Content (%) | No. of rRNAs | No. of tRNAs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Blastococcus sp. | TF02-8 | QOHK00000000 | 16,767,887 | 33 | 1026.0 | 3,982,980 | 380.9 | 3,814 | 75 | 8 | 47 |
| Blastococcus sp. | TF02A-30 | QOHJ00000000 | 15,562.207 | 38 | 922.0 | 4,129,003 | 466.8 | 4,008 | 74 | 6 | 48 |
| Blastococcus sp. | TF02-09 | QOHH00000000 | 9,367,969 | 37 | 558.0 | 4,132,992 | 297.8 | 3,953 | 73 | 11 | 47 |
| Blastococcus sp. | TBT05-19 | QOHI00000000 | 16,871,453 | 25 | 683.0 | 3,927,066 | 476.9 | 3,774 | 74 | 6 | 47 |
| Blastococcus sp. | TF02A-26 | QOHG00000000 | 12,236.063 | 54 | 627.0 | 4,678,378 | 217.9 | 4,561 | 74 | 6 | 47 |
| Blastococcus sp. | TF02A-35 | SPQP00000000 | 7,862,418 | 87 | 809.5 | 3,930,523 | 46.8 | 3,884 | 74 | 5 | 47 |
| Blastococcus sp. | CT_GayMR16 | SPQK00000000 | 5,162,206 | 47 | 157.2 | 4,520,567 | 136.3 | 4,472 | 73 | 8 | 47 |
| Blastococcus sp. | CT_GayMR19 | SPQL00000000 | 6,471,936 | 42 | 154.7 | 4,574,936 | 102.4 | 4,354 | 73 | 8 | 47 |
| Blastococcus sp. | CT_GayMR20 | SPQM00000000 | 1,759,527 | 345 | 37.1 | 5,475,077 | 37.1 | 5,501 | 73 | 7 | 56 |
| Geodermatophilus sp | DF01-2 | SPQN00000000 | 4,109,200 | 199 | 385.2 | 4,449,339 | 29.9 | 4,305 | 75 | 6 | 47 |
| Geodermatophilus sp | TF02-6 | QOHF00000000 | 12,613,686 | 53 | 639.0 | 4,725,362 | 162,9 | 4,448 | 75 | 7 | 49 |
Functional Assessment of Geodermatophilaceae Isolate Genomes
The genomes were analyzed for the Clusters of Orthrologous Groups (COG) functional categories to identify potential functionality of the isolates [24] by the use of the reCOGnizer tool workflow [25]. Functional profiling of the Geodermatophilaceae isolate genomes was also performed using PALADIN (version 1.4.2) with the raw genomic reads [26]. PALADIN detects open reading frames (ORFs) within the read data and converts them to protein sequences. Converted read protein sequences are aligned against a reference protein database using the Burrows-Wheeler Aligner [27]. PALADIN then assigns protein functions to the aligned proteins detected within the genome based on the reference database. Here, the UniRef90 database was used as the reference protein database [28]. Gene Ontology (GO) domains were assigned to each aligned genome protein sequence by parsing the UniProt report generated by PALADIN [24, 29]. The three GO domains are cellular component, molecular function, and biological process, and were used to assign broad functional categories to the isolate genomes.
Due to the potential novelty of the Geodermatophilaceae isolates, the genomes were evaluated for the production of secondary metabolites that could aid in the survival on stone surfaces (i.e. carotenoids) or that could have biotechnology or medical applications (i.e. antibiotics). The assembled and filtered contigs of each genome were used to determine potential secondary metabolite production through the bacterial version of antiSMASH version 5.0 [30].
Results and Discussion
Identification of Bacteria Isolated from Stone Surfaces
Several growth media were used to isolate a range of Actinobacteria, particularly members of the family Geodermatophilaceae, from stones. From the stones of the sampling regions, a total of 85 bacterial isolates were cultured, purified, and stored at -80oC.
A total of 40 bacteria were isolated from the stones collected from Tamil Nadu, India - 31 belonged to Actinobacteria (78%). Many of the isolated Actinobacteria belonged to the genera Geodermatophilus, Blastococcus, Mycobacterium, and Micrococcus. Nearly 90% of the Indian isolates were cultured from granite, while the rest were cultured from granodiorite. The 6 Blastococcus and 2 Geodermatophilus isolates were cultured from granite from several different sites (Table 1).
A total of 45 bacteria were isolated from New England stone samples - 25 belonged to Actinobacteria (56%). Prominent Actinobacteria cultured from New England stones included Dermacoccus, Arthrobacter, and Blastococcus. Other notable or unusual Actinobacteria included Auraticoccus, Micromonospora, and Branchiibius, among others. The Blastococcus isolates were cultured from the same built granite stone from Gay City, CT (Table 1).
Sanger Sequencing of the Complete 16S rRNA Gene of Geodermatophilaceae Isolates
Of the 85 bacteria isolated from the sampled stones, 11 were identified as belonging to the family Geodermatophilaceae. The full 16S rRNA gene of the 11 Geodermatophilaceae isolates was determined. The consensus sequences of the Geodermatophilaceae isolate 16S rRNA genes, including the top BLAST result and percent identity to each result, are summarized in Table S1. Two isolates belonged to the genus Geodermatophilus and 9 belonged to the genus Blastococcus.
Assembly of Geodermatophilaceae Isolate Genomes
The genomes of the 11 stone-dwelling isolates identified above as members of Geodermatophilaceae were shotgun sequenced. Assembly statistics and taxonomy assignments are summarized in Table 2. All isolate genomes were identified as belonging to the same genus as described by the full 16S rRNA gene sequence. Assembly lengths for Geodermatophilus genomes ranged from 4,451,532 to 4,725,362 base pairs, while the assembly lengths for Blastococcus genomes ranged in size from 3,927,160 to 5,476,194 base pairs. All genome assemblies were composed of less than 90 contigs, except for isolates DF01-2 and GayMR20, which contained 199 and 345 contigs, respectively. All genomes also had an N50 value of at least 30,000 base pairs. The average genome coverage was at least 230X for all genomes except for isolate GayMR20, which had approximately 80X average genome coverage. In addition, all isolates had a high G+C % value of 72% or higher, which is consistent with the high G+C % values found previously in Geodermatophilaceae isolates.
Assessment of the Novelty of Geodermatophilaceae Stone Isolates
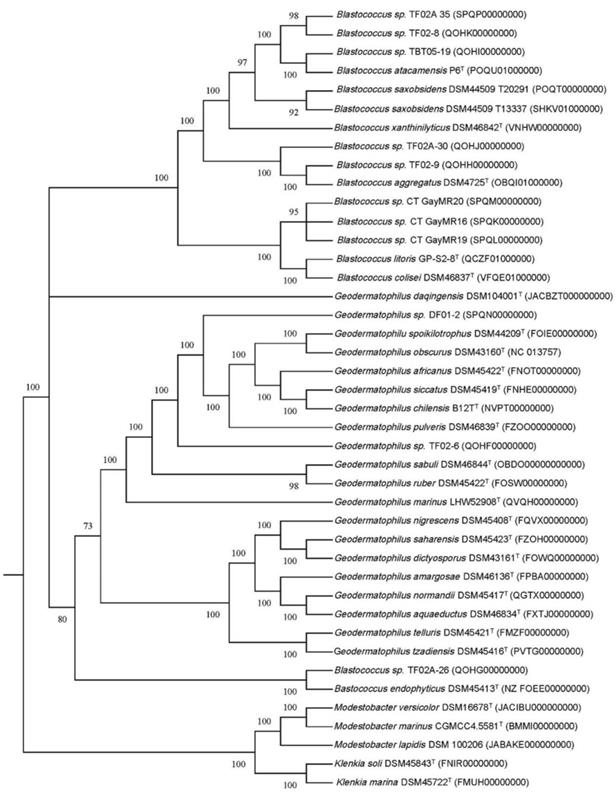
A maximum likelihood (ML) tree of the full 16S rRNA genes was constructed to determine the phylogeny of the 11 Geodermatophilaceae isolates (Fig. 1). Isolates DF01-2 and TF02-6 aligned near G. ruber and G. sabuli, but both were very distinct, indicating both as potential unique species. Similarly, all the Blastococcus isolates clustered several Blastococcus species, but were still distinct. Phylogenetic trees based on single genes are limited in scope. To obtain a better understanding of the phylogeny of the 11 isolates, a ML phylogenetic tree based on the entire genomes was constructed (Fig. 2). Phylogenetic analysis of the entire genomes confirmed 16 S rRNA gene phylogenetic tree and supports the idea that these isolates may represent potential novel species.
Maximum likelihood (ML) tree for the 16S rRNA sequences showing the position of the Geodermatophilaceae isolates. The tree consists of the following organisms and accession numbers in parenthesis Blastococcus sp. CT_GayMR20 (SPQM00000000); Blastococcus sp. CT_GayMR19 (SPQL00000000); Blastococcus sp. CT_GayMR16 (SPQK00000000); Blastococcus sp. TF02-9 (QOHH00000000); Blastococcus sp. TF02-8 MK239642; Blastococcus sp. TF02A-26 (QOHG00000000); Blastococcus sp. TF02A-30 (QOHJ00000000); Blastococcus sp. TF02A-35 (SPQP00000000);Blastococcus sp. TBT05-19 (QOHI00000000); Geodermatophilus sp. TF02-6 (QOHF00000000); Geodermatophilus sp. DF01-2 (SPQN00000000); Geodermatophilus africanus strain DSM 45422, isolate CF 11/1 (HE654550.1);Geodermatophilus chilensis strain B12TT (KX943328.2); Geodermatophilus normandii DSM:45417, type strain CF 5/3T (HE654546.1); Geodermatophilus arenarius type strain CF 5/4T (HE654547.1); Geodermatophilus daqingensis strain WT-2-1 (KX881378.1); Geodermatophilus tzadiensis DSM45416, type strain CF5/2T (HE654545.1);Geodermatophilus ruber DSM 45317, strain CPCC 201356 (EU438905); Geodermatophilus sabuli strain BMG 8133T (LN626269.1);Geodermatophilus aqueductis BMG801T DSM 46834 (LN626272); Geodermatophilus obscurus strain G20 DSM 43160 (CP001867); Geodermatophilus amargosae strain G96 DSM 46136 (HF679056; Geodermatophilus saharensis type strain CF5/5T (HE654551); Geodermatophilus dictyosporus, type strain G-5T (HF970584); Geodermatophilus nigrescens strain YIM 75980 (JN188947);geodermatophilus pulveris BMG825T (LN626270; Geodermatophilus poikilotrophus, type strain DSM 44209T (HF970583;Geodermatophilus siccatus strain DSM 45419, type strain CF6/1T (HE654548);Geodermatophilus marinus strain LHW52908 (MG200147); Klenkia marina, strain YIM M13156 T, DSM 45722 (LT746188); Klenkia soli strain PB34 16ST (JN033772.1); Klenkia terrae strain PB261 (JN033773): odestobacter lapidis strain MON3.1T (LN810544.1); Modestobacter lacusdianchii strain JXJ CY 19T (KP986567.1); Modestobacter multiseptatus strain AA826T (Y18646.1); Thalassiella azotivora strain DSD2 (KT630890);Nakamurella silvestris strain S20-107 (KP899234;); Blastococcus jejuensis strain KST3-10 (DQ200983); Blastococcus colisei strain BMG 822T (LN626273) ; Blastococcus litoris strain GP-S2-8T (MH128378); Blastococcus deserti strain SYSU D8006 (MH553383); Blastococcus aggregatus strain DSM 4725T (AJ430193.1); Blastococcus endophyticus strain YIM 68236T (GQ494034); Blastococcus capsensis sp. BMG 804T (LN626274); Blastococcus xanthinilyticus strain BMG 862T (LN626275); Blastococcus saxobsidens type strain DSM 44509T (FN600641); and Blastococcus atacamensis strain P6T (KX926540). The evolutionary history was inferred by using the Maximum Likelihood method and Tamura-Nei model [34]. The tree with the highest log likelihood (-7042.40) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura-Nei model, and then selecting the topology with superior log likelihood value. This analysis involved 47 nucleotide sequences. There were a total of 1570 positions in the final dataset. Evolutionary analyses were conducted in MEGA11 [35].

Tree inferred with FastME 2.1.6.1 [36] from Genome BLAST Distance Phylogeny approach (GBDP) distances calculated from genome sequences. The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values > 60 % from 100 replications, with an average branch support of 95.1 %. The tree was rooted at the midpoint [37] and redrawn in MEGA11 [35].
A whole genome-based taxonomic analysis via the Type (Strain) Genome Server (TYGS) platform [21] (https://tygs.dsmz.de) including digital DNA:DNA hybridization (dDDH) values [22] was performed to determine if these isolates represent new species (Fig. S1 and S2). The type-based species clustering using a 70% dDDH radius around each of the type strains was used as previously [31], while subspecies clustering was done using a 79% dDDH threshold as previously introduced [32]. These data suggest that all Blastococcus and Geodermatophilus isolates are potential novel species. Average nucleotide identity (ANI) analysis of these genomes (Fig. S3 and S4) confirmed that idea with ANI values well below the threshold of 95% for species delineation [33].
Functional Properties of Geodermatophilaceae Stone Isolates
Analysis of the 11 Geodermatophilaceae genomes for the number of genes associated with the Clusters of Orthrologous Groups (COG) functional categories showed that the pattern of distribution for each Blastococcus and Geodermatophilus isolate was like the patterns for B. saxobsidens DD2 and G. obscurus DSM 43160, respectively (Table S2 and S3).
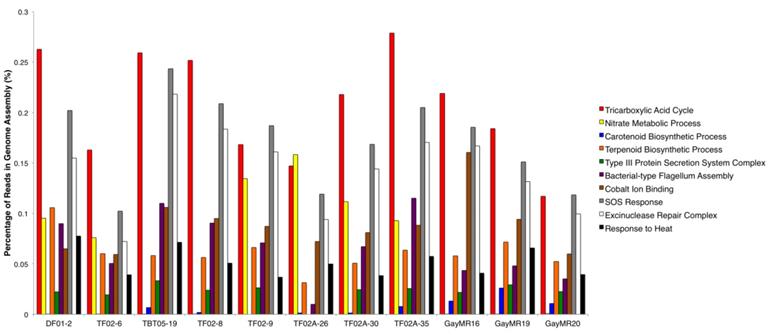
To further determine the functional capacity of the Geodermatophilaceae stone isolates, the raw genomic reads were analyzed using PALADIN. A total of 2,691 GO Terms were identified within the 11 genomes - 910 belonged to the 'Biological Process' GO term type, 1,638 belonged to the 'Molecular Function' GO term type, and 143 belonged to the 'Cellular Component' GO term type. Figure 3 summarizes 10 major GO terms that were prominent within each isolate genome and were relevant to survival on stone surfaces. Among these 10 functions, three functions that were in high abundance within all 11 genomes were the Tricarboxylic Acid Cycle (GO:0006099), SOS Response (GO:0009432), and the Excinuclease Repair Complex (GO: 0009380). Other functions that were enriched but in lower abundance in all 11 genomes include the Terpenoid Biosynthesis Process (GO:0016114), Bacterial-type Flagellum Assembly (GO: 0044780), Cobalt Ion Binding (GO:0050897), and Response to Heat (GO:0009408). Interestingly, the Type III Protein Secretion System Complex (GO:0030257) was the most abundant secretion system type in these genomes and was found in all 11 isolates except for Blastococcus isolate TF02A-26. The Nitrate Metabolic Process (GO:0042126) was another broad metabolic function that was present in high abundance in most of the isolate genomes but was completely absent from Blastococcus isolates TBT05-19, TF02-8, GayMR16, GayMR19, and GayMR20. The Carotenoid Biosynthetic Process (GO:0016117) was present in surprisingly low abundance within the isolate genomes, despite the highly pigmented morphology of most members of Geodermatophilaceae. This function was present at very low abundance within both Geodermatophilus isolates (DF01-2 and TF02-6), Blastococcus isolates TF02-8, TF02A-26, and TF02A-30. This function was also completely absent within Blastococcus isolate TF02-9.
Major Functions of Geodermatophilaceae Isolate Genomes. The relative abundances of 10 major functions identified within the 11 Geodermatophilaceae isolate genomes are summarized. Function abundances are reported as the percentage of genomic reads mapped to each function within each genome.
The antiSMASH version 5.0 program was also used on the assembled genomes of the 11 Geodermatophilaceae isolates to determine if the isolates had the potential to produce secondary metabolites, including antibiotics. The gene clusters detected in each isolate genome are summarized in Table 3. The Alkyl-O-Dihydrogeranyl-Methoxyhyrdoquinone biosynthesis gene cluster, under the Type 3 polyketide synthase (T3pks) metabolite type, was detected in every isolate genome. All isolate genomes also contained gene clusters associated with pigmentation production, in the forms of carotenoid or isorenierate biosynthesis. Many of these Geodermatophilaceae genomes also contained gene clusters associated with the production of antibacterial, antifungal, or even antiviral compounds, including stenothricin, pradimicin, nanchangmycin, istamycin, and fosfazinomycin. Interestingly, the Desferrioxamine B biosynthesis gene cluster, which is associated with siderophore iron-chelating activity, was detected in isolates TF02-8, TF02A-26, and TF02A-35. Several unknown secondary metabolites were also detected in isolates TF02-6, TF02-8, TF02-9, TF02A-30, TF02A-35, and GayMR20
Biosynthetic gene clusters for natural products found in the genomes from Geodermatophilacea.
| Bacterial species | Isolate | No. of Biosynthetic gene clusters 1 | NRPS 2 | PKS 3 | Terpene | Siderophore | Betalactone | Bacteriocin | Lanthipeptide |
|---|---|---|---|---|---|---|---|---|---|
| Blastococcus sp. | TF02-8 | 4 | 1 | 1 | 1 | 1 | |||
| Blastococcus sp. | TF02A-30 | 3 | 1 | 1 | 1 | ||||
| Blastococcus sp. | TF02-09 | 4 | 2 | 1 | 1 | ||||
| Blastococcus sp. | TBT05-19 | 3 | 1 | 1 | lassopeptide | ||||
| Blastococcus sp. | TF02A-26 | 1 | 1 | ||||||
| Blastococcus sp. | TF02A-35 | 5 | 1 | 1 | 1 | butyrolactone | 1 | ||
| Blastococcus sp. | CT_GayMR16 | 2 | 1 | 1 | |||||
| Blastococcus sp. | CT_GayMR19 | 3 | 1 | 1 | 1 | ||||
| Blastococcus sp. | CT_GayMR20 | 6 | 1 | 1 | 1 | 1 | indole | 1 | |
| Blastococcus saxobsidens | DD2 | 6 | 1 | 1 | 1 | 1 | lassopeptide | ||
| Geodermatophilus sp. | DF01-2 | 4 | 1 | 1 | 1 | NPSR-terpene hopene | |||
| Geodermatophilus sp. | TF02-6 | 5 | 1 | 2 | 1 | 1 | |||
| Geodermatophilus obscurus | DSM 43160 | 6 | 3 | 2 | 1 |
1Biosynthetic gene clusters were identified by the use of the antiSMASH software. 2NRPS: Nonribosomal peptide synthase. 3PKS: polyketide synthase including Type I, II, III, Trans-AT, and other types
In summary, we isolated 11 Geodermatophilaceae strains (9 Blastococcus and 2 Geodermatophilus isolates) and sequenced their genomes. These isolates represent potential novel species of these two bacterial genera. Analysis of their genomes revealed several unique traits that could play a role in their ecological niche.
Data availability. The draft genome sequences of these bacterial strains have been deposited in GenBank under the accession numbers listed in Table 2. Both the assembly and raw reads are available at DDBJ/ENA/GenBank under BioProject numbers: PRJNA478225, PRJNA478231 PRJNA478233, PRJNA478236, PRJNA478237, PRJNA478240, and PRJNA480027.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We like to thank the T3 course participants for their efforts on this project: Chhettri Saroja, Tsunemi Yamashita, Devin Thomas, Mohammad Alam, Rami M. Alroobi, Eric C. Atkinson, Nick Baer, Kayla Bieser, Nicolas Blouin, Louise J. Brogan, Jack Chen, Nicholas P. Edgington, Olivia L. George, Ghanshyam D. Heda, Amber Howerton, Jenna Luek, Paula Mazzer, Kelly Ann Miller, Daniel P. Moore, Shallee T. Page, Judith L. Roe, Kevin E. Shuman, and Kristy Townsend.
Partial funding and research support were provided from the University of New Hampshire CoRE program (LST, JB), the University of New Hampshire Summer Teaching Assistant Fellowship (NE), the University Grants Commission Raman Postdoctoral Fellowship of India (DD), The Binational Fulbright Commission in Jordan through the Jordanian Visiting Post-Doctoral Scholar Fellowship (S.M.A), Innovative Programs to Enhance Research Training (IPERT) from the National Institute of General Medical Sciences R25GM125674 (W.K.T.), New Hampshire-INBRE through an Institutional Development Award (IDeA), P20GM103506, from the National Institute of General Medical Sciences of the NIH (W.K.T.), and the College of Life Science and Agriculture at the University of New Hampshire-Durham. Sequencing was performed on an Illumina HiSeq2500 instrument purchased with NSF MRI grant DBI-1229361 (W.K.T.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cockell CS, Rettberg P, Horneck G, Wynn-Williams DD, Scherer K, Gugg-Helminger A. Influence of ice and snow covers on the UV exposure of terrestrial microbial communities: dosimetric studies. J Photoch Photobio B. 2002;68:23-32
2. Gtari M, Essoussi I, Maaoui R, Sghaier H, Boujmil R, Gury J. et al. Contrasted resistance of stone-dwelling Geodermatophilaceae species to stresses known to give rise to reactive oxygen species. Fems Microbiol Ecol. 2012;80:566-77
3. Louati M, Ennis NJ, Ghodhbane-Gtari F, Hezbri K, Sevigny JL, Fahnestock MF. et al. Elucidating the ecological networks in stone-dwelling microbiomes. Environmental Microbiology. 2020;22:1467-80
4. Gorbushina AA, Krumbein WE, Volkmann M. Rock surfaces as life indicators: New ways to demonstrate life and traces of former life. Astrobiology. 2002;2:203-13
5. Normand P, Daffonchio D, Gtari M. The family Geodermatophilaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The Prokaryotes. Berlin, Heidelberg: Springer. 2014 p. 361-79
6. Ennis NJ, Dharumaduri D, Bryce JG, Tisa LS. Metagenome Across a Geochemical Gradient of Indian Stone Ruins Found at Historic Sites in Tamil Nadu, India. Microb Ecol. 2021;81:385-95
7. Ennis NJ. Metagenomic analysis of the Microbial Communities Associated with Stone Surfaces: University of New Hampshire; 2018
8. Dulbecco R, Vogt M. Plaque Formation and Isolation of Pure Lines with Poliomyelitis Viruses. J Exp Med. 1954;99:167-82
9. Hunter-Cevera JC, Fonda ME, Belt A. Isolation of Cultures. In: Demain AL, Solomon NA, editors. Manual of Industrial Microbiology and Biotechnology. Washington, DC: American Society for Microbiology. 1986 p. 3-23
10. Luedeman.Gm. Geodermatophilus a New Genus of Dermatophilaceae (Actinomycetales). J Bacteriol. 1968;96:1848 -&
11. Reasoner DJ, Geldreich EE. A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol. 1985;49:1-7
12. Goodfellow M, Williams E. New Strategies for the Selective Isolation of Industrially Important Bacteria. Biotechnol Genet Eng. 1986;4:213-62
13. Murray MG, Thompson WF. Rapid Isolation of High Molecular-Weight Plant DNA. Nucleic Acids Res. 1980;8:4321-5
14. Cui XL, Mao PH, Zeng M, Li WJ, Zhang LP, Xu LH. et al. Streptimonospora salina gen. nov, sp nov, a new member of the family Nocardiopsaceae. Int J Syst Evol Micr. 2001;51:357-63
15. Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. Journal of molecular biology. 1975;94:441-8
16. Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci U S A. 1985;82:6955-9
17. Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J. et al. GenBank. Nucleic Acids Res. 2013;41:D36-D42
18. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20
19. Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A. et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. Journal of computational biology: a journal of computational molecular cell biology. 2013;20:714-37
20. Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614-24
21. Meier-Kolthoff JP, Goker M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nature communications. 2019;10:2182
22. Meier-Kolthoff JP, Auch AF, Klenk HP, Goker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. Bmc Bioinformatics. 2013;14:60
23. Richter M, Rossello-Mora R, Oliver Glockner F, Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32:929-31
24. Galperin MY, Wolf YI, Makarova KS, Alvarez RV, Landsman D, Koonin EV. COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021;49:D274-D81
25. Sequeira JC, Rocha M, Alves MM, Salvador AF. UPIMAPI, reCOGnizer and KEGGCharter: Bioinformatics tools for functional annotation and visualization of (meta)-omics datasets. Comput Struct Biotec. 2022;20:1798-810
26. Westbrook A, Ramsdell J, Schuelke T, Normington L, Bergeron RD, Thomas WK. et al. PALADIN: protein alignment for functional profiling whole metagenome shotgun data. Bioinformatics. 2017;33:1473-8
27. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
28. Suzek BE, Wang YQ, Huang HZ, McGarvey PB, Wu CH, Consortium U. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31:926-32
29. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM. et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25-9
30. Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Lee SY. et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019;47:W81-W7
31. Liu Y, Lai Q, Goker M, Meier-Kolthoff JP, Wang M, Sun Y. et al. Genomic insights into the taxonomic status of the Bacillus cereus group. Scientific reports. 2015;5:14082
32. Meier-Kolthoff JP, Hahnke RL, Petersen J, Scheuner C, Michael V, Fiebig A. et al. Complete genome sequence of DSM 30083(T), the type strain (U5/41(T)) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand Genomic Sci. 2014 9
33. Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Micr. 2007;57:81-91
34. Tamura K, Nei M. Estimation of the Number of Nucleotide Substitutions in the Control Region of Mitochondrial-DNA in Humans and Chimpanzees. Mol Biol Evol. 1993;10:512-26
35. Tamura K, Stecher G, Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol. 2021;38:3022-7
36. Lefort V, Desper R, Gascuel O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol Biol Evol. 2015;32:2798-800
37. Farris JS. Estimating Phylogenetic Trees from Distance Matrices. Am Nat. 1972;106:645-67
Author contact
![]() Corresponding author: Mailing address: Department of Molecular, Cellular, and Biomedical Sciences, University of New Hampshire, 46 College Rd., Durham, NH 03824-2617. Email: louis.tisaedu Telephone: 1-603-862-2442 Fax: 1-603-862-2621.
Corresponding author: Mailing address: Department of Molecular, Cellular, and Biomedical Sciences, University of New Hampshire, 46 College Rd., Durham, NH 03824-2617. Email: louis.tisaedu Telephone: 1-603-862-2442 Fax: 1-603-862-2621.