ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2024; 12:44-46. doi:10.7150/jgen.92255 This volume Cite
Short Research Paper
Draft genome sequencing and assembly of Favolaschia claudopus CIRM-BRFM 2984 isolated from oak limbs
David Navarro1,2 ![]() , Elodie Drula1,3, Delphine Chaduli1,2, Robert Cazenave4, Steven Ahrendt5, Jie Wang5, Anna Lipzen5, Chris Daum5, Kerrie Barry5, Igor V. Grigoriev5,6, Anne Favel1,2, Marie-Noëlle Rosso1, Francis Martin7,8
, Elodie Drula1,3, Delphine Chaduli1,2, Robert Cazenave4, Steven Ahrendt5, Jie Wang5, Anna Lipzen5, Chris Daum5, Kerrie Barry5, Igor V. Grigoriev5,6, Anne Favel1,2, Marie-Noëlle Rosso1, Francis Martin7,8
1. INRAE, Aix Marseille Univ, UMR1163 Biodiversité et Biotechnologie Fongiques, 13288, Marseille, France.
2. CIRM-CF, INRAE, Aix Marseille Univ, UMR 1163, 13009, Marseille, France.
3. AFMB, CNRS, Aix Marseille Univ, UMR 7257, USC 1408, 13009, Marseille, France.
4. Association Mycologique de Bigorre, 65600, Séméac, France.
5. U.S. Department of Energy Joint Genome Institute, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
6. Department of Plant and Microbial Biology, University of California Berkeley, Berkeley, CA 94720, USA.
7. INRAE, Univ de Lorraine, UMR 1136 Interactions Arbres/Microorganismes, 54280, Champenoux, France.
8. Institute of Applied Mycology, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, Hubei 430070, China.
Received 2023-11-15; Accepted 2024-1-8; Published 2024-2-17
Abstract

Favolaschia claudopus, a wood-inhabiting basidiomycete of the Mycenaceae family, is considered an invasive species that has recently spread from Oceania to Europe. The CIRM-BRFM 2984 strain of this fungus was originally isolated from a basidiome collected from the fallen limb of a decayed oak tree in Southwest France. The genome sequence of this strain shared characteristics with other Mycenaceae species, including a large genome size and enriched content of protein-coding genes. The genome sequence provided here will facilitate further investigation on the factors that contribute to the successful global dissemination of F. claudopus.
Keywords: Favolaschia, genome, Mycenaceae, decayed wood, Agaricales, Invasive species
Introduction
The genus Favolaschia belongs to the Mycenaceae family within the order Agaricales. It is known for its poroid basidiomes that are found in dead plant materials worldwide. Previously considered as a variety of Favolaschia calocera, F. claudopus (Singer) Q.Y. Zhang & C. Dai has recently been elevated to the species status (1). F. claudopus is a wood-inhabiting fungus classified as an invasive species that has recently spread from Oceania to Europe (2; 3; 1). It is suspected that the introduction of this species may be favored by the presence of black locust (Robinia pseudoacacia), which is the primary host of F. claudopus. The adaptive capabilities of this species have raised concerns among mycologists as they could potentially endanger local fungal species (4; 5). The first specimens of F. claudopus in metropolitan France were observed in Pyrénées-Atlantiques (southwest France). We have carried out genome sequencing of the F. claudopus strain CIRM-BRFM 2984 as part of the 1000 Fungal Genomes (1KFG) project, an extensive international research initiative focused on sequencing the genomes of various fungal taxa, in collaboration with the Joint Genome Institute of the U.S. Department of Energy (6).
CIRM-BRFM 2984, a strain of F. claudopus, was isolated from a basidiome collected from the fallen branch of a decayed Quercus tree in Agnos, France, in November 2018 (refer to Fig. 1 for visual representation). The basidiome was dried and preserved using voucher number AMB49273. The isolation process involved transferring tissues from the basidiome to agar plates containing malt extract supplemented with antibiotics (0.025% chloramphenicol and 0.04% gentamicin). Successive subcultures without antibiotics were performed to verify the absence of contaminants. To confirm the identity of the isolated strain, the rDNA barcoding region ITS1-5,8S-ITS2 was PCR-amplified and sequenced, as previously described (7). Following molecular authentication, the strain was deposited in the Biological Resource Center CIRM-CF (International Center of Microbial Resources, Marseille, France; https://doi.org/10.15454/KJQW-SJ57; www.cirm-fungi.fr) under the accession number CIRM-BRFM 2984.
Favolaschia claudopus basidiomes.

Genomic DNA was extracted from the ground mycelia according to the protocol described by Lomascolo et al. (8). In order to enhance gene structural annotation, we sequenced the mRNAs obtained after a 3-day growth period on three different media: A: Glucose 10 g.L-1, bactopeptone 5 g.L-1, yeast extract 2.5 g.L-1; B: Malt extract 15 g.L-1, salts (KH2PO4 0.2 g.L-1, CaCl2·2H2O 1.32.10-2 g.L-1, MgSO4·7H2O 0.5 g.L-1, FeSO4·7H2O 0.07 g.L-1, ZnSO4·7H2O 7.77.10-3 g.L-1, MnSO4·H2O 3.63.10-3 g.L-1, CuSO4·5H2O 7.2.10-4 g.L-1) and thiamine 250.10-3 g.L-1; C: Maltose 10 g.L-1, yeast extract 2.5 g.L-1, salts and thiamine 250.10-3 g.L-1. Fresh mycelia were harvested, pooled, and ground in liquid nitrogen using a cryogenic grinder (SPEX Sample Prep; UK). Subsequently, total RNAs were extracted from 100 mg of ground tissue using TRIZOL (Ambion), followed by precipitation with isopropanol, resuspension in water, and treatment with RNase-free Dnase I (QIAGEN). Total RNAs were then precipitated with LiCl and resuspended in DEPC-treated water according to the protocol described by Miyauchi et al. (9).
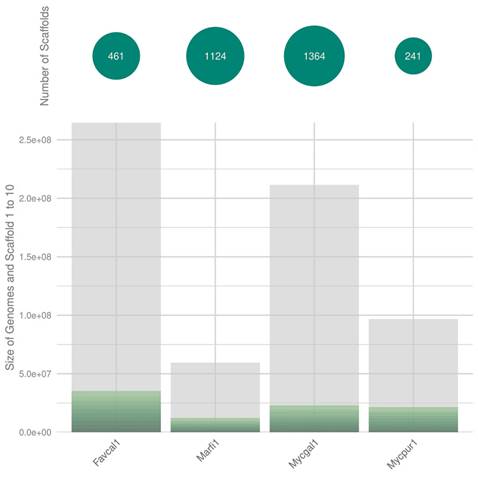
The F. claudopus CIRM-BRFM 2984 v1.0 genome was sequenced from 162 µg of genomic DNA using the Pacific Biosciences sequencing platform (>10kb PacBio libraries with Blue Pippin size selection), assembled with Falcon v. 0.0.8 (10), polished with Arrow version SMRTLINK v8.0.0.80529 (https://www.pacb.com/support/software-downloads), and annotated with the JGI Annotation Pipeline (6). mRNA sequences were obtained using Illumina RNA-Seq data assembled with Trinity v2.11.0 (11). The genome size of F. claudopus (265 Mbp) was similar to that of other strains of the Mycenaceae family (9). The draft genome assembly (Table 1) was significantly improved compared to that of previously sequenced Mycena species, with the 10 largest scaffolds covering 13.3% of the genome (Fig. 2). We used BUSCO (BUSCO v3.0.2) with default parameters and the agaricales_odb10 database (12) to benchmark universal single-copy orthologues and assess the completeness of the genome Favolaschia claudopus CIRM-BRFM 2984 v1.0). The completeness of these genome assembly was compared to that of three Mycenaceae genomes; Marasmius fiardii PR-910 v1.0, Mycena galopus ATCC-62051 v1.0 and Mycena pura 9144 v1.0. The BUSCO scores varied from 87.4% to 92.8%. Regarding the genome of Favolaschia claudopus, the completeness was the highest (92.8%) and only 6.4% of BUSCO genes were missing. It should be noticed that 86.8% (3358) of the BUSCO genes were identified as duplicated sequences. After annotation of the protein-coding genes, we identified a large repertoire of CAZymes (1,340 genes), peptidases (891 genes), and transporters (1,592 genes).
Genome assembly information.
| Genome Assembly size | 264.58Mbp |
|---|---|
| Sequencing read coverage depth | 20.47x |
| # of contigs | 461 |
| Contig N50/L50 | 61/1.46Mbp |
| # of gene models | 49,883 |
Genome sequence assembly. The size of the 10 largest scaffolds (green sections) from F. claudopus CIRM-BRFM 2984 v1.0 (Favcal1_2) was compared with that of the genomes available for the related species Marasmius fiardii PR-910 v1.0 (Marfi1), Mycena galopus ATCC-62051 v1.0 (Mycgal1) and Mycena pura (Mycpur1).
Further research, including comparative genomic analysis with other Mycena species, will aid in determining whether the expansive genome of F. claudopus has facilitated its ability to disperse and establish itself in diverse ecological niches.
Acknowledgements
Work (proposal: 10.46936/10.25585/60001060) conducted by the U.S. Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility, was supported by the Office of Science of the U.S. Department of Energy operated under Contract No. DE-AC02-05CH11231. This work was also supported by the Laboratory of Excellence ARBRE (ANR-11-LABX-0002-01) (to FM).
Data availability
The genome assembly and annotations are available from The JGI Fungal Genomics Portal MycoCosm (6; https://mycocosm.jgi.doe.gov/Favcal1_2) and the Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JAWWNJ000000000. The version described in this paper is version JAWWNJ010000000. Biosample and Bioproject have been deposited at GenBank under the accession number SAMN37908076 and PRJNA1030398 respectively.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang QY, Dai YC. Taxonomy and Phylogeny of the Favolaschia calocera Complex (Mycenaceae) with Descriptions of Four New Species. Forests. 2021;12(10):1397
2. Vizzini A, Zotti M. Favolaschia calocera, a tropical species collected in Italy. Mycotaxon -Ithaca Ny-. 2002 1; 82:169-76
3. Vizzini A, Zotti M, Mello A. Alien fungal species distribution: the study case of Favolaschia calocera. Biol Invasions. 2009 1; 11(2):417-29
4. Ainsworth AM, Farley D, Gainey P, Penna P, Suz LM. Invasion of the orange ping-pong bats: the rapidly changing distribution of Favolaschia calocera. Field Mycology. 2015;16(4):113-202
5. Chuzho K, Dkhar MS. Ecological Determinants of Wood-Rotting Fungal Diversity and First Report of Favolaschia calocera, an Invasive Species from India. Proc Natl Acad Sci, India, Sect B Biol Sci. 2019 1; 89(4):1177-88
6. Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R. et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42(Database issue):D699-704
7. Navarro D, Chaduli D, Taussac S, Lesage-Meessen L, Grisel S, Haon M. et al. Large-scale phenotyping of 1,000 fungal strains for the degradation of non-natural, industrial compounds. Commun Biol. 2021 15; 4(1):1-10
8. Lomascolo A, Cayol JL, Roche M, Guo L, Robert JL, Record E. et al. Molecular clustering of Pycnoporus strains from various geographic origins and isolation of monokaryotic strains for laccase hyperproduction. Mycological Research. 2002 1; 106(10):1193-203
9. Miyauchi S, Kiss E, Kuo A, Drula E, Kohler A, Sánchez-García M. et al. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat Commun. 2020 12;11(1):5125
10. Chin CS, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods. 2016;13(12):1050-4
11. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644-52
12. Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015 1; 31(19):3210-2
Author contact
![]() Corresponding author: David Navarro; david.navarrofr.
Corresponding author: David Navarro; david.navarrofr.