ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2026; 14:10-17. doi:10.7150/jgen.130158 This volume Cite
Research Paper
Draft Genomes of Geographically Distinct Strains and Progeny of the Ectomycorrhizal Basidiomycete Laccaria bicolor
Francis M. Martin1 ![]() , Emmanuelle Morin1, Alan Kuo2, Igor Miquel1, Jessy Labbé3, François Le Tacon1, Laure Fauchery1, Annegret Kohler1, William Andreopoulos2, Alex Copeland2, Hui Sun2*, Asaf Salamov2, Anna Lipzen2, James Han2, Kurt LaButti2, Andrew Tritt2, Kerry Barry2, Igor V. Grigoriev2,4
, Emmanuelle Morin1, Alan Kuo2, Igor Miquel1, Jessy Labbé3, François Le Tacon1, Laure Fauchery1, Annegret Kohler1, William Andreopoulos2, Alex Copeland2, Hui Sun2*, Asaf Salamov2, Anna Lipzen2, James Han2, Kurt LaButti2, Andrew Tritt2, Kerry Barry2, Igor V. Grigoriev2,4
1. Université de Lorraine, INRAE, UMR Interactions Arbres/Microorganismes, INRAE-Grand Est-Nancy, 54280 Champenoux, France.
2. U.S. Department of Energy Joint Genome Institute, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
3. Biosciences Division, Oak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, TN 37831, USA.
4. Department of Plant and Microbial Biology, University of California Berkeley, Berkeley, CA 94720, USA.
* deceased.
Received 2025-12-18; Accepted 2026-1-9; Published 2026-1-30
Abstract

The ectomycorrhizal fungus Laccaria bicolor is a key symbiotic mutualist in forest ecosystems, where it enhances nutrient uptake and promotes the growth of host trees. Here, we present genome assemblies of 14 geographically distinct strains and progeny of L. bicolor, providing new insights into the intraspecific genomic diversity. Pangenome analysis revealed substantial variation in assembly size (42-96 Mbp), gene content (16,084-26,800 genes), and single nucleotide polymorphism (SNP) density (0.04-12.08 SNPs/kb). This variation likely reflects genuine biological differences among strains adapted to diverse environmental conditions, although differences in assembly quality and repeat content may also play a role. These genomic resources, comprising draft genome assemblies with comprehensive annotations, will facilitate comparative studies of the genetic diversity and functional traits underlying the ecological success of this model ectomycorrhizal fungus.
Keywords: Ectomycorrhizal fungi, Laccaria bicolor, pangenome, comparative genomics, genetic diversity, symbiosis
Introduction
Ectomycorrhizal fungi form mutualistic associations with most tree species in forest ecosystems and play essential roles in nutrient cycling and tree growth [1]. Laccaria bicolor, a member of the Hydnangiaceae family within the order Agaricales (Basidiomycota), has long served as an experimental model in mycorrhizal research [2]. The genome of L. bicolor strain S238N-H82 comprises a 60.7-megabase assembly containing approximately 23,130 predicted protein-coding genes [3]. Analysis of this genome and associated transcriptomes has revealed gene networks involved in symbiosis development, including effector-type secreted proteins known as mycorrhiza-induced small secreted proteins (MiSSPs), which play critical roles in symbiotic establishment [4,5].
Notably, the L. bicolor genome encodes a reduced number of carbohydrate-active enzymes (CAZymes) involved in plant cell wall degradation while retaining the ability to degrade microbial cell wall polysaccharides. This pattern reflects the dual saprotrophic and biotrophic lifestyles characteristic of ectomycorrhizal fungi [3], and the loss of genes encoding plant cell wall-degrading enzymes has been confirmed across dozens of ectomycorrhizal species [6].
Understanding intraspecific variability in genomic features is essential for comprehensive comparative and evolutionary genomic analyses of ectomycorrhizal fungi [7]. Analysis of the L. bicolor pangenome and its intraspecific genetic variability can provide crucial insights into the genomic diversity, adaptation, and evolution of this ecologically important, symbiotic fungus. Our pangenomic analysis identified core genes shared among geographically distinct strains and genes shared among the progeny of the L. bicolor S238N lineage. We also characterised accessory genes that may facilitate local adaptation to diverse environments.
Materials and Methods
Fungal Strains and Culture Conditions
Strains D101 and S238O of L. bicolor were collected in Quebec Province, Canada (no known host), whereas strain DR170 was collected in the Upper Peninsula of Michigan, USA, beneath Pinus resinosa. Strain S238N originated from a basidiocarp collected beneath Tsuga mertensiana at Crater Lake, Oregon, USA, and subsequently produced basidiocarps with Douglas fir seedlings in the INRAE-Nancy greenhouse. Strains 81306, Cham3, and N203 were collected from Douglas fir plantations at Barbaroux, Saint-Brisson, and Chammet in the Auvergne region.
The S238N-H82, S238N-H70, and S238N-H53 strains are sib-monokaryons derived from the parental strain S238N. The S238N-H82 × H70 strain was generated by crossing the homokaryotic strains S238N-H70 and S238N-H82. Strain S238N 93.12 originated from L. bicolor S238N after 20 years of storage in liquid N2. L. bicolor CBS 594.89 (origin unknown) and CBS 559.96 (collected under Pinus sylvestris in the Netherlands) were obtained from the CBS fungal collection (Westerdijk Institute, Utrecht, NL).
Free-living vegetative mycelium of each L. bicolor strain was grown for approximately three weeks in Pachlewski medium [8] at 25°C on agar. Mycelial colony edges were sampled, snap-frozen in liquid N2, and ground into a fine powder.
DNA Extraction, Genome Sequencing, and Assembly
Total genomic DNA was extracted from three grams of vegetative mycelium using a CTAB-based protocol [3] and purified using Qiagen genomic-tip 500/G columns, following the manufacturer's instructions [3]. All new L. bicolor genomes and transcriptomes were sequenced using Illumina technology, with some genomes additionally combined with PacBio sequencing technology.
For transcriptomes, stranded cDNA libraries were generated using the Illumina TruSeq Stranded RNA LT Kit. mRNA was purified from one µg of total RNA using magnetic beads containing poly T oligonucleotides, fragmented, and reverse-transcribed using random hexamers and SSII (Invitrogen), followed by second-strand synthesis. Fragmented cDNA was treated with end-repair, A-tailing, adapter ligation, and eight cycles of PCR amplification.
For all genomes, Illumina regular fragment libraries were produced from 100 ng of DNA sheared to 300 bp using a Covaris LE220 (Covaris) and size-selected using SPRI beads (Beckman Coulter). Fragments were treated with end-repair, A-tailing, and ligation of Illumina-compatible adapters (IDT, Inc.) using the KAPA-Illumina library creation kit (KAPA Biosystems).
Additionally, for L. bicolor 81306, D101, DR170, N203, S238O, S238N, S238N-H82, S238N-H70, and S238N-H53, Illumina 4 kb Long-Mate Pair (LMP) CLRS libraries were produced from 5 µg of DNA sheared using a Covaris g-TUBE (Covaris) and gel size-selected for 4 kb. The sheared DNA was treated with end repair and ligated to biotinylated adapters containing loxP. Adapter-ligated DNA fragments were circularised via recombination using a Cre excision reaction (NEB). Circularised DNA templates were randomly sheared using the Covaris LE220 (Covaris) and treated with end repair and A-tailing using the KAPA-Illumina library creation kit (KAPA Biosystems), followed by immobilisation of mate pair fragments on streptavidin beads (Invitrogen). Illumina-compatible adapters (IDT, Inc.) were ligated to the mate pair fragments, and 10-12 cycles of PCR were used to enrich the final library (KAPA Biosystems).
Both genome and transcriptome libraries were quantified using KAPA Biosystem's next-generation sequencing library qPCR kit (Roche) and run on a Roche LightCycler 480 real-time PCR instrument. Quantified libraries were multiplexed, and pooled libraries were prepared for sequencing on the Illumina HiSeq platform using a TruSeq paired-end cluster kit, v3 or v4, with Illumina's cBot instrument to generate the clustered flow cells. Sequencing was performed on Illumina HiSeq 2000 or 2500 sequencers using HiSeq TruSeq SBS sequencing kits, v3 or v4, following a 2×150 bp (2×100 bp for LMP) indexed run recipe.
RNA-Seq reads were filtered and trimmed for contamination and quality assessment. Using BBDuk (https://sourceforge.net/projects/bbmap/), raw reads were evaluated for artefact sequences by k-mer matching (k-mer = 25), allowing one mismatch, and the detected artefacts were trimmed from the 3' end. RNA spike-in reads, PhiX reads, and reads containing Ns were also removed. Quality trimming was performed using the Phred trimming method, which was set at Q6. Following trimming, reads below the length threshold were removed (minimum length 25 bases or one-third of the original read length, whichever was longer). Filtered reads were assembled into consensus sequences using Trinity ver. 2.1.1.
All DNA reads filtered for artefact/process contamination were assembled with AllPathsLG versions R41043, R44849, R47710, and R49403 [9]. For CBS 594.89, CBS 559.96, S238N-H82×H70, and Cham3, which lacked LMP data, Velvet [10] assemblies were generated and used to produce in silico LMP libraries with inserts of 3000 ± 300 bp size.
The genomes of strains D101, DR170, N203, S238N 93.12, S238N-H82, and S238N-H70 were also improved using PacBio sequencing. Unamplified libraries were generated using the Pacific Biosciences standard template preparation protocol for creating >10 kb libraries (>20 kb for S238N 93.12 and 3 kb for DR170). Five µg of gDNA was used for each library preparation. DNA was sheared using Covaris g-TUBEs to generate fragments >10 kb (3 kb for DR170). Sheared DNA fragments were prepared using the Pacific Biosciences SMRTbell Template Preparation Kit. The fragments were treated with DNA damage repair, the ends were repaired to be blunt-ended and 5' phosphorylated, and Pacific Biosciences hairpin adapters were ligated to create the SMRTbell templates for sequencing. Templates were purified using exonuclease treatments and size-selected using AMPure PB beads or the Sage Sciences Blue Pippin Size-Selection system for S238N 93.12. PacBio sequencing primers were annealed to the SMRTbell template library, and Version P4 or P6 sequencing polymerase was bound. The prepared SMRTbell template libraries were sequenced on a Pacific Biosciences RSII sequencer using Version C2 or C4 chemistry and 1×120 or 1×240 sequencing movie runtime.
AllPathsLG assemblies were patched using error-corrected PacBio data with PBJelly v12.9.14 [11] and polished with Quiver version smrtanalysis_2.3.0.140936. p5 (https://github.com/PacificBiosciences/GenomicConsensus/). For S238N 93.12, PacBio data were filtered with smrtanalysis and assembled with Falcon v.20150310 (https://github.com/PacificBiosciences/FALCON), and improved with finisherSC v. 1.0 [12] and polished with Quiver.
Gene Prediction and Annotation
Gene models were predicted using the JGI Annotation Pipeline, which integrates multiple gene prediction algorithms with RNA-seq evidence [13]. Functional annotation was performed by comparing the predicted proteins with the Pfam, KOG, and KEGG databases. Secreted proteins were predicted in the genomes of L. bicolor strains using a custom bioinformatic pipeline [15].
Orthology and Pangenome Analysis
Orthology of the protein repertoires was assessed using FastOrtho/Silix (50% identity, 50% coverage) to identify core genes (present in all strains), dispensable genes (present in at least two strains), and strain-specific genes. Strain-specific genes were further compared with all fungal genomes at JGI to identify species-specific genes.
Phylogenomic Analysis
Phylogenomic analysis was performed as described by [3]. Briefly, we identified clusters containing single-copy genes, aligned each cluster with MAFFT, eliminated ambiguous regions (containing gaps and poorly aligned regions), and concatenated the single-gene alignments with GBLOCKS. We constructed a phylogenetic tree using RAxML with the standard algorithm, the PROTGAMMAWAG model of sequence evolution, and 1000 bootstrap replicates. The final species tree was estimated using ASTRAL [14], with 2,038 unrooted gene trees.
SNP Prediction
Single nucleotide polymorphisms (SNPs) were predicted for 16 strains relative to the reference genome (Laccaria bicolor v2.0) using BWA-MEM for read mapping and SAMtools/BCFtools for variant calling, as described previously [7]. The repeated regions were not masked. A phylogenetic tree was generated using RAxML 7.7.2 with the GTRGAMMA model and 500 bootstrap replicates.
Results
Genome Assembly and Annotation Statistics
The genome assembly sizes of L. bicolor strains ranged from 42.12 to 96.43 Mbp (Table 1). The number of predicted genes ranged from 16,084 to 26,800, with average protein lengths between 310 and 397 amino acids across all strains. Pfam domains were identified in 6,192-14,251 genes per strain (Table 2). This considerable variation in assembly size and gene number likely reflects the differences in assembly quality, repeat content, and genuine biological variation among strains.
Summary statistics of genome assemblies
| Strain / assembly version | Assembly (Mbp) | Contigs # | N50 | L50 (Kbp) | Scaffolds # | N50 (#) | L50 (Kbp) | Repeat (%) | GC (%) |
|---|---|---|---|---|---|---|---|---|---|
| Laccaria bicolor S238N-H82 v2.0 | 60.71 | 584 | 50 | 342.8 | 55 | 5 | 3262.84 | 24.80 | 46.95 |
| Laccaria bicolor S238N-H82 v1.0 | 64.88 | 4399 | 180 | 71.0 | 665 | 21 | 786.28 | 21.20 | 46.97 |
| Laccaria bicolor S238N v1.0 | 67.66 | 6556 | 636 | 21.3 | 1577 | 113 | 160.33 | 21.68 | 47.27 |
| Laccaria bicolor S238N 93.12 v1.1 * | 96.43 | 1034 | 109 | 236.9 | 1034 | 109 | 236.92 | 22.86 | 46.84 |
| Laccaria bicolor S238N-H82xH70 v1.0 | 42.12 | 4436 | 404 | 24.1 | 3788 | 332 | 28.50 | 14.13 | 47.90 |
| Laccaria bicolor S238N-H70 v1.0 | 57.05 | 2269 | 133 | 107.9 | 1564 | 96 | 150.93 | 22.86 | 47.34 |
| Laccaria bicolor S238N-H53 v1.0 | 51.78 | 3014 | 219 | 57.3 | 1284 | 88 | 157.00 | 20.70 | 47.49 |
| Laccaria bicolor DR170 v1.0 | 79.07 | 7156 | 756 | 29.1 | 1941 | 185 | 107.45 | 25.97 | 47.00 |
| Laccaria bicolor S238O v1.0 | 57.06 | 3633 | 214 | 56.1 | 1370 | 68 | 195.42 | 22.20 | 47.31 |
| Laccaria bicolor D101 v1.0 | 70.03 | 4003 | 257 | 62.0 | 2920 | 171 | 106.84 | 22.37 | 47.31 |
| Laccaria bicolor N203 v1.0 | 69.63 | 2759 | 184 | 80.3 | 1866 | 109 | 143.29 | 25.25 | 47.03 |
| Laccaria bicolor Cham3 v1.0 | 44.65 | 3687 | 353 | 30.2 | 3327 | 338 | 32.50 | 15.67 | 47.71 |
| Laccaria bicolor 81306 v1.0 | 50.95 | 2780 | 209 | 57.7 | 946 | 74 | 194.83 | 18.47 | 47.51 |
| Laccaria bicolor CBS 594.89 v1.0 | 69.98 | 4627 | 309 | 50.6 | 4530 | 299 | 52.42 | 8.59 | 48.44 |
| Laccaria bicolor CBS 559.96 v1.0 | 43.46 | 3844 | 373 | 27.4 | 3721 | 364 | 28.44 | 15.28 | 47.71 |
| Laccaria amethystina LaAM-08-1 v2.0 | 52.58 | 1999 | 151 | 89.3 | 1299 | 112 | 121.59 | 21.88 | 46.62 |
| Agaricus bisporus var bisporus H97 v2.0 | 30.20 | 254 | 35 | 262.5 | 29 | 6 | 2334.61 | 2.19 | 46.48 |
| Amanita muscaria Koide v1.0 | 40.70 | 3814 | 266 | 30.0 | 1101 | 54 | 145.60 | 6.08 | 47.55 |
* Sequencing and assembly of the dikaryotic strain
Summary statistics for the annotated genomes
| Strain | # of genes | avg. exon length (nt) | avg. intron length (nt) | Exons per gene | avg. protein length (aa) | # genes w/ Pfam domain | BUSCO score* |
|---|---|---|---|---|---|---|---|
| Laccaria bicolor S238N-H82 v2.0 | 23132 | 220 | 92 | 5.28 | 356 | 6920 | 96.5 |
| Laccaria bicolor S238N-H82 v1.0 | 20614 | 210 | 93 | 5.4 | 367 | NA | 96.4 |
| Laccaria bicolor S238N v1.0 | 21724 | 227 | 80 | 5.04 | 351 | 6876 | 95.9 |
| Laccaria bicolor S238N-H82xH70 v1.0 | 17045 | 230 | 75 | 5.28 | 372 | 6192 | 96.7 |
| Laccaria bicolor S238N-H70 v1.0 | 19903 | 224 | 83 | 5.31 | 365 | 6681 | 95.1 |
| Laccaria bicolor S238N-H53 v1.0 | 18468 | 230 | 76 | 5.29 | 373 | 6437 | 96.8 |
| Laccaria bicolor DR170 v1.0 | 24004 | 198 | 78 | 5.33 | 310 | 8193 | 85.7 |
| Laccaria bicolor S238O v1.0 | 17767 | 220 | 79 | 5.29 | 388 | 6514 | 95.8 |
| Laccaria bicolor D101 v1.0 | 22538 | 237 | 86 | 5.02 | 360 | 7177 | 92.1 |
| Laccaria bicolor N203 v1.0 | 21909 | 213 | 86 | 5.33 | 343 | 6720 | 94.1 |
| Laccaria bicolor Cham3 v1.0 | 16084 | 217 | 74 | 5.32 | 386 | 6192 | 96.2 |
| Laccaria bicolor 81306 v1.0 | 17791 | 234 | 81 | 5.34 | 378 | 6413 | 99.7 |
| Laccaria bicolor CBS 594.89 v1.0 | 26800 | 224 | 70 | 5.70 | 397 | 14251 | 94.3 |
| Laccaria bicolor CBS 559.96 v1.0 | 16977 | 236 | 69 | 5.28 | 371 | 7631 | 97.0 |
| Laccaria amethystina LaAM-08-1 v2.0 | 17553 | 242 | 70 | 5.12 | 364 | 6174 | 97.7 |
| Agaricus bisporus var bisporus H97 v2.0 | 10432 | 232 | 72 | 6.05 | 426 | 5359 | 98.0 |
| Amanita muscaria Koide v1.0 | 18153 | 253 | 73 | 4.54 | 328 | 6244 | 97.0 |
* BUSCO Agaricomycetes gene set
Pangenome Analysis and Gene Orthology
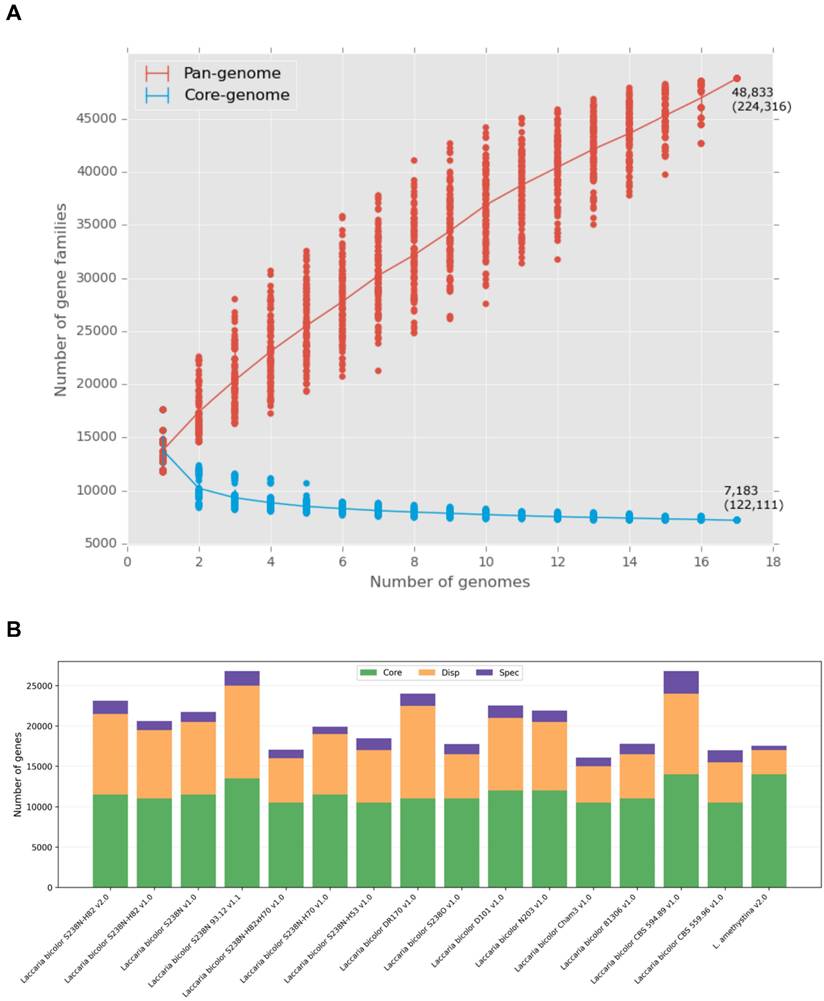
Pangenome analysis revealed substantial genetic diversity among the 14 L. bicolor strains (Figure 1A). The core genes (present in all strains) ranged from 10,516 to 14,568 per strain, the dispensable genes ranged from 2,937 to 10,361, and the strain-specific genes ranged from 228 to 3,708 (Figure 1B). When including the related species L. amethystina, the pangenome encompassed 48,833 gene families, whereas the core genome stabilised at approximately 7,183 gene families. The substantial number of dispensable and strain-specific genes highlights the functional diversity within L. bicolor, potentially enabling adaptation to different soil and climatic conditions.
Core and pangenomes of Laccaria bicolor. (A) Accumulation curves showing how the pangenome size (red) increases and the core genome size (blue) decreases with each additional genome. (B) Distribution of core (green), dispensable (orange), and strain-specific (purple) genes across the strains. Stacked bar chart representation.
Phylogenomic Relationships
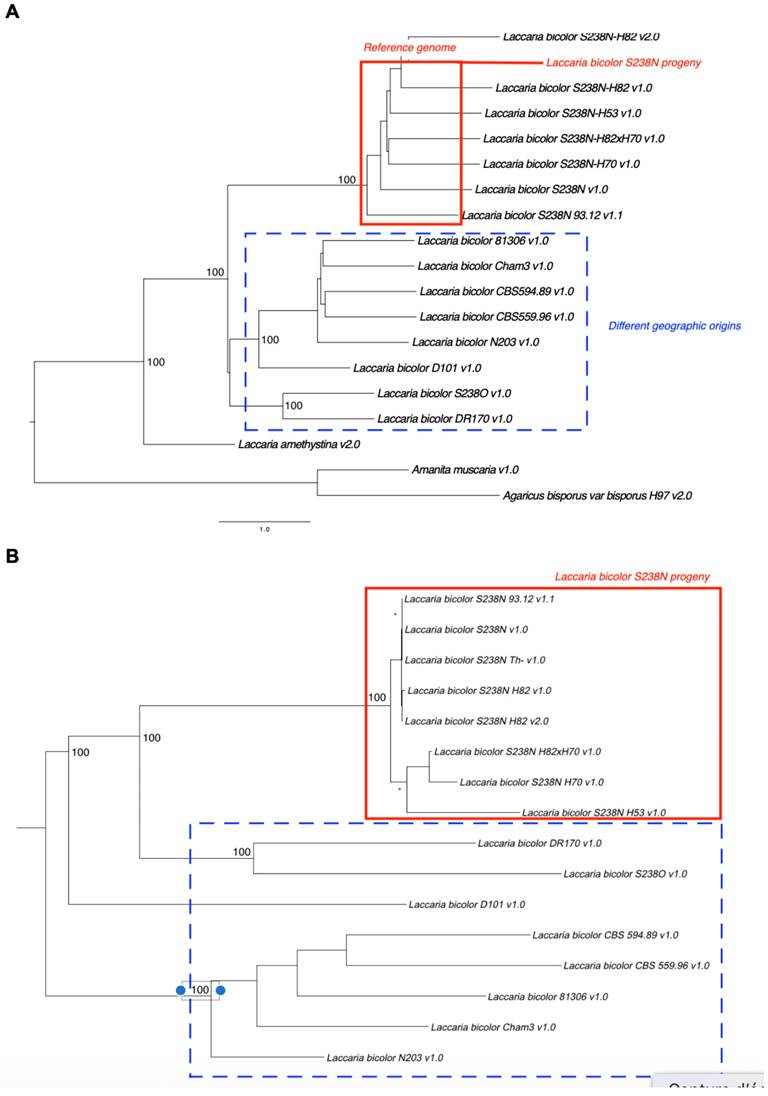
Phylogenomic analysis based on 2,038 single-copy orthologues clearly distinguished L. bicolor strains from L. amethystina, as well as from the more distantly related Agaricales, Agaricus bisporus and Amanita muscaria (Figure 2A). Several distinct clades were evident within L. bicolor. The parental strain S238N and its progeny (homokaryotic and dikaryotic strains) formed a well-supported, monophyletic group. Other distinct clusters indicate the geographic structuring of genetic diversity within species.
(A) Phylogenomic tree of Laccaria isolates based on 2,038 single-copy orthologous gene trees inferred using ASTRAL. The tree included 16 L. bicolor strains, one L. amethystina strain, and two outgroup species (Agaricus bisporus and Amanita muscaria). Branch support values are shown at the major nodes. (B) Maximum likelihood phylogeny of L. bicolor strains based on genome-wide SNP data, constructed using RAxML with the GTRGAMMA model and 500 bootstrap replicates.
Single Nucleotide Polymorphism Analysis
SNP analysis revealed substantial variations among the strains (Figure 2B). SNP density ranged from 3.15 SNPs/kb (S238N-H82 v.1.0 vs. S238N-H82 v2.0) to 12.08 SNPs/kb (DR170 vs. S238N-H82 v2.0). The proportion of SNPs located within the coding regions varied between 44.57% and 55.34%. The SNP-based phylogeny closely corresponded to the phylogenomic tree derived from conserved proteins (Figure 2A).
The substantial sequence divergence (>1%) observed in certain strains, such as L. bicolor DR170, likely reflects geographic isolation and the structuring of genetic diversity within the species. Such levels of divergence raise questions about whether some strains warrant recognition as distinct taxonomic entities.
Secretome Characteristics
All strains possessed substantial complements of genes encoding secreted carbohydrate-active enzymes (CAZymes; 111-278 genes), lipases (8-17), proteases (40-97), and small secreted proteins (371-606). Complete annotations are available at the Laccaria pangenome portal of MycoCosm. Variations in the number of secreted proteins may reflect strain-specific adaptations to different hosts and environmental conditions.
Discussion
This study presents a comprehensive pangenomic analysis of Laccaria bicolor, substantially expanding the genomic resources available for this model ectomycorrhizal species. Our analysis revealed that L. bicolor possesses a large accessory genome, with approximately 7,183 core gene families and up to 48,833 total gene families. This open pangenome structure is consistent with species inhabiting diverse environments and associated with multiple host species.
The substantial variation in genome size (42-96 Mbp) and gene content (16,084-26,800 genes) among strains suggests genuine biological differences, although assembly quality may contribute to some of this variation. Strain CBS 594.89, with the largest genome (69.98 Mbp) and highest gene count (26,800), may represent a strain with expanded gene families and elevated transposable element activities.
SNP analysis revealed varying levels of divergence among strains, indicating that our collection captured both recent derivatives of common laboratory strains and geographically isolated populations. The variation in small secreted protein (SSP) repertoires (371-606 genes) was particularly noteworthy. Given the importance of specific MiSSPs in symbiotic establishment [4,5,16,17], strain-specific SSP repertoires may reflect adaptations to different host species or environmental conditions. A detailed functional enrichment analysis of core, accessory, and strain-specific genes, including CAZyme and MiSSP distribution across the pangenome, will be addressed in future studies.
These genomic resources will enable future studies on comparative effector biology, local adaptation, population genomics, functional validation of symbiosis-related genes, and synthetic community. This study contributes to the growing body of fungal pangenome studies and demonstrates the value of population-scale genomics in understanding the ecology and evolution of mycorrhizal fungi. The substantial genetic diversity within L. bicolor suggests considerable potential for adaptation to changing environmental conditions, with implications for conservation strategies and biotechnological applications, including reforestation efforts and optimisation of plant-fungal partnerships for sustainable forestry.
Acknowledgements
This material is based on work (project: 10.46936/10.25585/60000842) conducted by the U.S. Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy, operated under contract no. DE-AC02-05CH11231. This research was also supported by the Laboratory of Excellence ARBRE (ANR-11-LABX-0002-01), the Genomic Science Program (Plant-Microbe Interactions project) funded by the US Department of Energy, Office of Science, Biological and Environmental Research (contract DE-AC05-00OR22725) (to J.L.), and the Lorraine Region Council (to F.M.M.). We thank Dr. Claire Veneault-Fourrey and Aurélie Deveau for their helpful discussions. The Word-plugin Paperpal was used for language editing.
Data Availability
All genome assemblies and annotations are publicly available through the JGI MycoCosm portal (http://mycocosm.jgi.doe.gov) at: https://mycocosm.jgi.doe.gov/Laccaria. Raw sequencing reads were deposited in the NCBI Sequence Read Archive (BioProject # PRJNA82349, PRJNA372751, PRJNA372752, PRJNA337346, PRJNA333959, PRJNA258987, PRJNA200600, PRJNA410717, PRJNA234844, PRJNA333972, PRJNA403323, PRJNA258985, PRJNA258990, PRJNA258983, PRJNA243647, PRJNA258982).
Competing Interests
The authors have declared that no competing interests exist.
References
1. Martin F, van Der Heijden MA. The mycorrhizal symbiosis: research frontiers in genomics, ecology, and agricultural application. New Phytol. 2024;242:1486-1506
2. Martin F, Selosse MA. The Laccaria genome: a symbiont blueprint decoded. New Phytol. 2008;180:296-310
3. Martin F, Aerts A, Ahrén D. et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature. 2008;452:88-92
4. Plett JM, Kemppainen M, Kale SD. et al. A secreted effector protein of Laccaria bicolor is required for symbiosis development. Curr Biol. 2011;21:1197-1203
5. Marqués-Gálvez JE, Pandharikar G, Basso V, Kohler A, Lackus ND, Barry K, Keymanesh K, Johnson J, Singan V, Grigoriev IV, Vilgalys R, Martin F, Veneault-Fourrey C. Populus MYC2 orchestrates root transcriptional reprogramming of defence pathway to impair Laccaria bicolor ectomycorrhizal development. New Phytol. 2024;242:658-674
6. Miyauchi S, Kiss E, Kuo A. et al. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat Commun. 2020;11:5125
7. Dauphin B, Freitas Pereira M, Kohler A. et al. Cryptic genetic structure and copy-number variation in the ubiquitous forest symbiotic fungus Cenococcum geophilum. Environ Microbiol. 2021;23:6536-6556
8. Pachlewski R, Pachlewska J. Studies on symbiotic properties of mycorrhizal fungi of pine (Pinus sylvestris L.) with the aid of the method of mycorrhizal synthesis in pure cultures on agar. Warsaw, Poland: Forest Research Institute; 1974
9. Gnerre S, Maccallum I, Przybylski D. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA. 2011;108:1513-1518
10. Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821-829
11. English AC, Richards S, Han Y. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE. 2012;7:e47768
12. Lam K-K, LaButti K, Khalak A. et al. FinisherSC: a repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics. 2015;31:3207-3209
13. Grigoriev IV. et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42:D699-D704
14. Mirarab S, Reaz R, Bayzid MS. et al. ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics. 2014;30:i541-i548
15. Pellegrin C, Morin E, Martin FM, Veneault-Fourrey C. Comparative analysis of secretomes from ectomycorrhizal fungi with an emphasis on small-secreted proteins. Front Microbiol. 2015;6:1278
16. Plett JM, Daguerre Y, Wittulsky S. et al. Effector MiSSP7 of the mutualistic fungus Laccaria bicolor stabilizes the Populus JAZ6 protein and represses jasmonic acid (JA) responsive genes. Proc Natl Acad Sci USA. 2014;111:8299-8304
17. Kang H, Chen X, Kemppainen M. et al. The small secreted effector protein MiSSP7.7 of Laccaria bicolor is required for the establishment of ectomycorrhizal symbiosis. Environ Microbiol. 2020;22:1435-1446
Author contact
![]() Corresponding author: Francis M. Martin, E-mail: francis.martinfr.
Corresponding author: Francis M. Martin, E-mail: francis.martinfr.