ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2015; 3:72-74. doi:10.7150/jgen.12574 This volume Cite
Short Research Communication
Antibiotic Resistant and Virulence Determinants of Staphylococcus haemolyticus C10A as Revealed by Whole Genome Sequencing
Kok-Gan Chan1 ![]() , Kim Tien Ng2, Teik Min Chong1, Yong Kek Pang2, Adeeba Kamarulzaman2, Wai-Fong Yin1, Kok Keng Tee2
, Kim Tien Ng2, Teik Min Chong1, Yong Kek Pang2, Adeeba Kamarulzaman2, Wai-Fong Yin1, Kok Keng Tee2
1. Division of Genetics and Molecular Biology, Institute of Biological Sciences, Faculty of Science, University of Malaya, Kuala Lumpur 50603, Malaysia.
2. Faculty of Medicine, University of Malaya, Kuala Lumpur 50603, Malaysia.
Published 2015-6-3
Abstract
Staphylococcus haemolyticus is one of the pathogens that harbor a high level of antibiotic resistance. Here, we highlighted the potential determinants for multidrug resistance and virulence from the draft genome of Staphylococcus haemolyticus strain C10A, isolated from a patient with chronic obstructive pulmonary disease exacerbation.
Keywords: Staphylococcus haemolyticus, whole genome sequencing, multidrug resistance, chronic obstructive pulmonary disease (COPD).
Chronic obstructive pulmonary disease (COPD), a chronic obstruction of lung airflow, is commonly associated with episodes of acute deterioration termed as "exacerbation" (1). Nearly 50% of COPD are due to viral infections, while the remaining are associated with bacterial infections (2, 3). Antibiotics are commonly prescribed for COPD exacerbations (4). As such, the emergence of multidrug resistant bacteria is possible, particularly in patients with recurrent COPD exacerbations.
Phenotypic assessment of resistance against commonly used antibiotics is widely practiced. However, such assessment is incapable of detecting the in vivo potential of the bacteria to escape the effect of antibiotics (5, 6). Recent advancement in next generation sequencing technologies has deemed to be a convenient and cost effective approach to assess the genomic properties of organisms (7). Here, based on genomic data generated from whole genome sequencing (WGS), antimicrobial resistance profile of S. haemolyticus strain C10A, isolated from the sputum of a 40-year-old male COPD subject with 20 years of smoking history was investigated in silico. During recruitment, the ratio of forced expiratory volume to forced viral capacity (FEV1/FVC) was less than 70% and the spirometry indicated a less than 12% of improvement of FEV1 after the bronchodilator test.
Isolation of the bacteria was performed using tryptic soy agar at 37°C followed by phenotypic identification using Microflex MALDI-TOF (Bruker Daltonik GmbH, Leipzig, Germany) bench-top mass spectrometer. Classification of bacteria was then performed using the built-in Bruker MALDI Biotyper Real Time Classification software (Version 3.1, Build 65). Genomic DNA of strain C10A was extracted using MasterPureTM DNA Purification Kit (Epicentre Inc., Madison, WI, USA). Illumina MiSeq sequencing platform (Illumina, Inc., CA) was employed to sequence the DNA library prepared using Nextera XT DNA sample preparation kit (Illumina, Inc., CA). The acquired sequence data was then subjected to quality trimming and de novo assembly with CLC Genomic Workbench (version 7.0). Sequence reads with low quality (<Q20), ambiguous nucleotides and sequence length less than 50 nucleotides were discarded (8). For de novo assembly, contigs of at least 500 bp and 30-fold coverage were selected. The assembled reads were subsequently subjected to annotation using NCBI prokaryotic annotation pipeline (PGAAP) in search of putative genes coding for antibiotic resistance.
With the spectra generated from mass spectrometry, the closest match of strain C10A was S. haemolyticus with the score value of 2.107 indicating secure genus and probable species identification. To gain further evidence of the bacterial identification, the closest neighbor displayed by RAST server (9) was S. haemolyticus JCSC1435, a multidrug resistant strain exhibiting high genomic plasticity mediated by numerous insertion sequences (10).
The draft genome of S. haemolyticus C10A was 2,430,312 bp in size, with an average G+C content of 32.6%. Sequence assembly produced a total of 87 contigs with average size of 27,632 bp and maximum contig size of 153,180 bp. The sequencing depth was at 74.1-fold. Results from PGAAP revealed a total of 2,387 coding sequences with 5 rRNAs and 57 tRNAs identified in the genome. The associated bioproject number for S. haemolyticus C10A genome is PRJNA246625.
In search of genes associated with resistance towards antimicrobial agents, the annotation revealed the presence of candidates against a wide spectrum of antibiotics including aminoglycosides, macrolides, quinolones, penicillins, tetracyclines and glycopeptides in the genome of S. haemolyticus strain C10A. Figure 1 demonstrated the clustering of these identified determinants together with a series of possible virulence factors including components of cvfC, heme oxygenases and peptidase U32, with other S. haemolyticus and Staphylococcus strains.
Phylogenetic analysis of putative genes associated with antibiotics resistance and virulence in the genome of S. haemolyticus strain C10A. Multiple sequence alignments was performed using ClustalW and evolutionary history was deduced using Maximum Likelihood method based on JTT matrix-based model. Triangles (▲) at each branches indicates the putative genes identified in the genome of S. haemolyticus strain C10A.
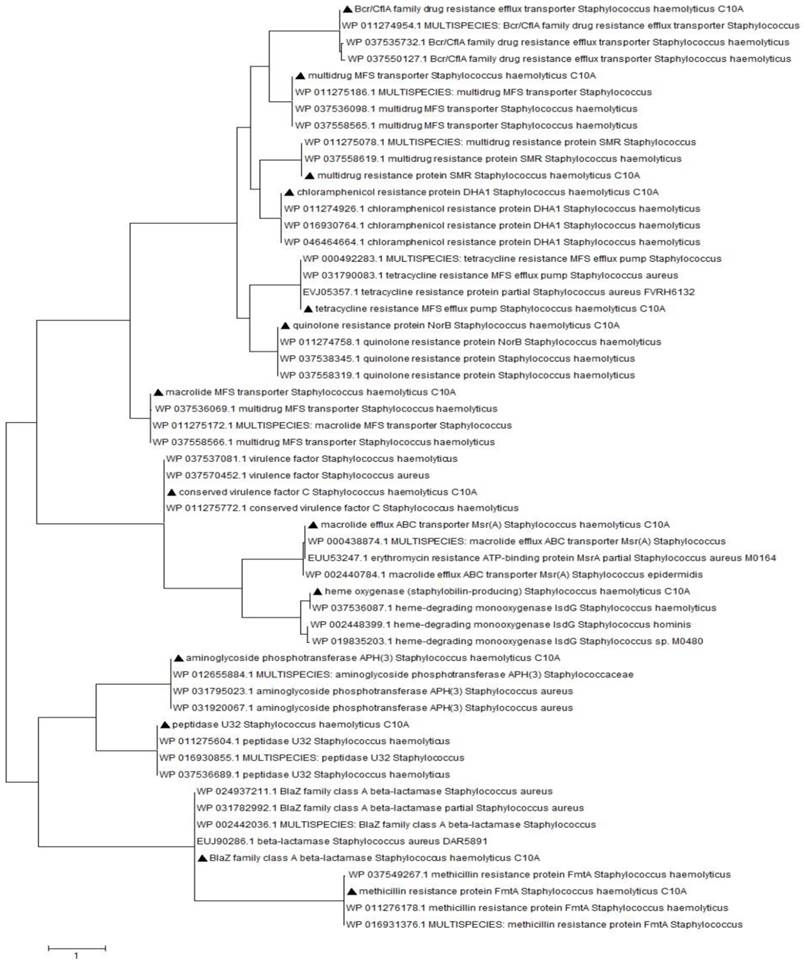
Genome sequencing and annotation of S. haemolyticus strain C10A in the present study has demonstrated the concise detection of multiple antibiotic resistance and virulence genes. Such observation has postulated the necessity to weigh the risk of antibiotic resistance, prior to its prescription for COPD patients.
Nucleotide sequence accession number: The genome sequence of S. haemolyticus C10A isolate has been deposited in GenBank under accession no. JPRW01000000. The version described in this paper is the first version.
Acknowledgements
This research was financed by the University of Malaya High Impact Research (HIR) Grants (UM-MOHE HIR Grant UM.C/625/1/HIR/MOHE/CHAN/14/1, no. H-50001-A000027; UM-MOHE HIR Grant UM.C/625/1/HIR/MOHE/CHAN/01, No. A000001-50001) to Kok-Gan Chan and UM-MOHE HIR Grant UM.C/625/1/HIR/MOHE/CHAN/2, no. H-50001-A000012 to Kok Keng Tee which are gratefully acknowledged.
Ethic Statements
The study was approved by the University Malaya Medical Centre (UMMC) Medical Ethics Committee. Standard, multilingual consent forms validated by the Medical Ethics Committee were used. Written consent was obtained from all study participants prior to collection of specimen.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mackay AJ, Hurst JR. COPD exacerbations: causes, prevention, and treatment. Med Clin North Am. 2012;96:789-809
2. Sethi S. Molecular diagnosis of respiratory tract infection in acute exacerbations of chronic obstructive pulmonary disease. Clin Infect Dis. 2011;52(Suppl 4):S290-295
3. Sykes A, Mallia P, Johnston SL. Diagnosis of pathogens in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2007;4:642-646
4. Miravitlles M, Espinosa C, Fernandez-Laso E, Martos JA, Maldonado JA, Gallego M. Relationship between bacterial flora in sputum and functional impairment in patients with acute exacerbations of COPD. Study Group of Bacterial Infection in COPD. Chest. 1999;116:40-46
5. Garcha D, Donaldson G, McHugh T, Wedzicha J, Thurston S. Impact of antibiotic exposure on the development of antibiotic resistance in COPD patients. Microbiology. 2013;1:2
6. Thurston SJ, Donaldson GC, McHugh TD, Wedzicha JA. Impact of COPD severity and sputum production on antibiotic resistance. Thorax. 2011;66(Suppl 4):A11-A11
7. Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet. 2012;13:601-612
8. Chong TM, Yin WF, Mondy S, Grandclement C, Dessaux Y, Chan KG. Heavy-metal resistance of a France vineyard soil bacterium, Pseudomonas mendocina strain S5.2, revealed by whole-genome sequencing. J Bacteriol. 2012;194:6366
9. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA. et al. The RAST Server: rapid annotations using subsystems technology. BMC genomics. 2008;9:75
10. Takeuchi F, Watanabe S, Baba T, Yuzawa H, Ito T, Morimoto Y, Kuroda M, Cui L, Takahashi M, Ankai A, Baba S, Fukui S, Lee JC, Hiramatsu K. Whole-genome sequencing of Staphylococcus haemolyticus uncovers the extreme plasticity of its genome and the evolution of human-colonizing staphylococcal species. J Bacteriol. 2005;187:7292-7308
Figures
Author contact
![]() Corresponding author: Division of Genetics and Molecular Biology, Institute of Biological Sciences, Faculty of Science, University of Malaya, Kuala Lumpur 50603, Malaysia; E-mail: kokganedu.my
Corresponding author: Division of Genetics and Molecular Biology, Institute of Biological Sciences, Faculty of Science, University of Malaya, Kuala Lumpur 50603, Malaysia; E-mail: kokganedu.my