ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2015; 3:82-84. doi:10.7150/jgen.12886 This volume Cite
Short Research Communication
Draft Genome Sequences of Two Species of “Difficult-to-Identify” Human-Pathogenic Corynebacteria: Implications for Better Identification Tests
Luis G. C. Pacheco1, ![]() , Ana L. Mattos-Guaraldi2, Carolina S. Santos1, Adonney A. O. Veras3, Luis C. Guimarães3, 4, Vinícius Abreu4, Felipe L. Pereira5, Siomar C. Soares5, Fernanda A. Dorella5, Alex F. Carvalho5, Carlos G. Leal5, Henrique C. P. Figueiredo5, Juliana N. Ramos2, 6, Veronica V. Vieira6, Eric Farfour7, Nicole Guiso7, Raphael Hirata Jr.2, Vasco Azevedo4, Artur Silva3, #, Rommel T. J. Ramos3, #,
, Ana L. Mattos-Guaraldi2, Carolina S. Santos1, Adonney A. O. Veras3, Luis C. Guimarães3, 4, Vinícius Abreu4, Felipe L. Pereira5, Siomar C. Soares5, Fernanda A. Dorella5, Alex F. Carvalho5, Carlos G. Leal5, Henrique C. P. Figueiredo5, Juliana N. Ramos2, 6, Veronica V. Vieira6, Eric Farfour7, Nicole Guiso7, Raphael Hirata Jr.2, Vasco Azevedo4, Artur Silva3, #, Rommel T. J. Ramos3, #, ![]()
1. Institute of Health Sciences, Federal University of Bahia (UFBA), Salvador, BA, Brazil.
2. Faculty of Medical Sciences, Rio de Janeiro State University (UERJ), Rio de Janeiro, RJ, Brazil.
3. Institute of Biological Sciences, Federal University of Pará (UFPA), Belém, PA, Brazil.
4. Institute of Biological Sciences, Federal University of Minas Gerais (UFMG), Belo Horizonte, MG, Brazil.
5. National Reference Laboratory for Aquatic Animal Diseases, Ministry of Fisheries and Aquaculture, Federal University of Minas Gerais (UFMG), Belo Horizonte, MG, Brazil.
6. National Institute for Quality Control in Health - Oswaldo Cruz Foundation (INCQS - Fiocruz), Rio de Janeiro, RJ, Brazil.
7. Unité de Prévention et Thérapie Moléculaires des Maladies Humaines, Institut Pasteur, Paris, France.
# These authors share credit for senior authorship in this work.
Published 2015-8-1
Abstract
Non-diphtheriae Corynebacterium species have been increasingly recognized as the causative agents of infections in humans. Differential identification of these bacteria in the clinical microbiology laboratory by the most commonly used biochemical tests is challenging, and normally requires additional molecular methods. Herein, we present the annotated draft genome sequences of two isolates of “difficult-to-identify” human-pathogenic corynebacterial species: C. xerosis and C. minutissimum. The genome sequences of ca. 2.7 Mbp, with a mean number of 2,580 protein encoding genes, were also compared with the publicly available genome sequences of strains of C. amycolatum and C. striatum. These results will aid the exploration of novel biochemical reactions to improve existing identification tests as well as the development of more accurate molecular identification methods through detection of species-specific target genes for isolate's identification or drug susceptibility profiling.
Keywords: Corynebacterium spp., Emerging pathogens, Biochemical tests, Molecular identification
Non-diphtheriae Corynebacteria are usually found as constituents of the normal microbiota of the human skin and mucosae. However, several Corynebacterium species have been increasingly implicated as the causative agents of opportunistic and nosocomial infections in recent years. These emerging human pathogenic corynebacteria are often regarded as “difficult-to-identify” and multidrug resistance is a common finding among isolates of the most frequently recovered species (1-3).
Corynebacterium minutissimum, which is the causative agent of the chronic skin condition erythrasma, has emerged in recent years as an important causative agent of invasive infections and of surgical site infections, both in immunocompromised patients and in immunocompetent individuals (1, 4). The species Corynebacterium xerosis is also a frequently reported human pathogen, with isolates being identified in cases that include ear infections, brain abscesses and osteomyelitis (2). This latter species is of particular interest due to the zoonotic potential and due to the difficulties associated to identification of this microorganism in the clinical microbiology laboratory by the most traditionally employed tests (1, 2, 5, 6).
In this report, we announce the draft genome sequences of two isolates of emerging human pathogenic Corynebacterium species, namely: (i) C. minutissimum 1941, isolated from an hospitalized cancer patient (1); and (ii) C. xerosis ATCC 373T, a type strain of the species that was isolated from the ear discharge of a child. At the time of preparation of this work, no other genome assembly was publicly available for the species C. xerosis.
Both isolates rendered identification scores higher than 2.0 when analyzed by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) using MALDI-Biotyper v.2.0 software, as described previously (7). Analysis of 16S ribosomal RNA was also concordant with identifications obtained by MALDI-TOF MS and by conventional biochemical methods.
Genomic DNA of the isolates was extracted using the QIAamp DNA minikit (Qiagen) protocol and DNA sequencing was performed with the 318 chip of the Ion Torrent PGM platform (Life Technologies), using fragment libraries. De novo assembly of the sequences into contigs was achieved using MIRA (8) and SPAdes (9) assemblers, and curation to reduce the gaps was done with the Lasergene v.11 Suite (DNASTAR). Sequence assembly produced a total of 42 contigs for C. minutissimum 1941 and 152 contigs for C. xerosis ATCC 373T. The draft genome sequence of the former species had an estimated length of 2,780,059, with a G+C content of 61.1%. The latter species showed an estimated genome size of 2,749,912, with a G+C content of 68.7%. Automatic annotation using the RAST server (10) allowed for identification of 2,547 protein encoding genes (PEGs) and 64 RNA genes in the C. minutissimum 1941 genome; the number of PEGs in the C. xerosis ATCC 373T genome was 2,613, with 56 RNA genes.
The availability of these whole genome sequences now provides the opportunity to evaluate novel biochemical reactions to improve identification tests currently used in clinical microbiology laboratories. We used the SEED annotation environment (http://www.theseed.org/) to investigate the genomic basis for some typical biochemical features commonly used to differentiate these “difficult-to-identify” corynebacterial species. We also included in these comparisons the publicly available genomic sequences for C. striatum ATCC 6940 (ACGE00000000) and C. amycolatum SK46 (NZ_ABZU00000000); this latter species frequently generates ambiguous identifications mostly with C. xerosis and C. striatum. The absence of genes coding for enzymes involved in maltose utilization, such as maltose phosphorylase (EC 2.4.1.8), was a distinctive characteristic of C. striatum ATCC 6940 in comparison to the other species. On the other hand, potential activity of respiratory nitrate reductase (EC 1.7.99.4) was only detected in this isolate along with C. xerosis ATCC 373T, but not in C. amycolatum SK46 or C. minutissimum 1941. Additionally, potential for galactose uptake and utilization was also not detected in C. minutissimum 1941.
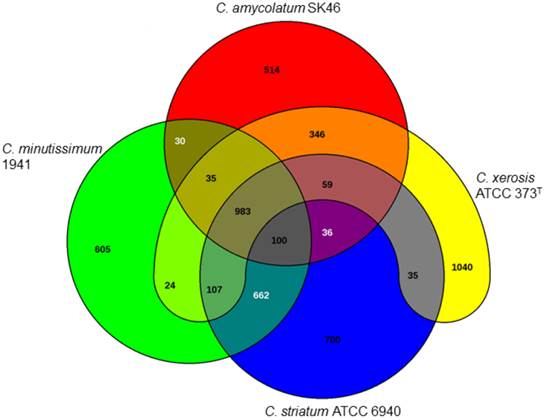
Furthermore, whole genome sequencing may also aid development of more accurate molecular diagnostic methods through detection of species-specific target genes for isolate's identification or drug susceptibility profiling. Analysis with the EDGAR server (edgar.computational.bio.uni-giessen.de) allowed for detection of 983 CDSs shared by the four isolates evaluated in this study. Most importantly, it was possible to detect a number of distinctive genes for each species, when compared to each other: C. xerosis (1,040 CDSs); C. striatum (700 CDSs); C. minutissimum (605 CDSs); and C. amycolatum (514 CDSs) (Fig. 1).
Venn diagram demonstrating the numbers of coding sequences shared by the four Corynebacterium species analyzed in this study.
Nucleotide sequence accession numbers. These Whole Genome Shotgun projects have been deposited at DDBJ/EMBL/GenBank under the accessions LAYS00000000 (C. xerosis ATCC 373T) and LAYQ00000000 (C. minutissimum 1941). The versions described in this paper are versions LAYS01000000 and LAYQ01000000.
Acknowledgements
This work was supported by grants from the Brazilian Research Funding Agencies CNPq, CAPES (PROCAD 071/2013), and FAPESB (JCB0031/2013).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Martins C, Faria L, Souza M, Camello T, Velasco E, Hirata R Jr, Thuler L, Mattos-Guaraldi A. Microbiological and host features associated with corynebacteriosis in cancer patients: a five-year study. Mem Inst Oswaldo Cruz. 2009;104:905-913
2. Bernard K. The genus corynebacterium and other medically relevant coryneform-like bacteria. J Clin Microbiol. 2012;50:3152-3158
3. Ramana KV, Vikram G, PadmaWali P, K A, Rao M, Rao SD, MS RM, CH V, Rao R. Non Diphtheritic Corynebacteria (NDC) and Their Clinical Significance: Clinical Microbiologist's Perspective. Am J Epidemiol Infect Dis. 2014;2:83-87
4. Shin JY, Lee WK, Seo YH, Park YS. Postoperative Abdominal Infection Caused by Corynebacterium minutissimum. Infect Chemother. 2014;46:261-263
5. Funke G, Lawson PA, Bernard KA, Collins MD. Most Corynebacterium xerosis strains identified in the routine clinical laboratory correspond to Corynebacterium amycolatum. J Clin Microbiol. 1996;34:1124-1128
6. Boschert V, Berger A, Konrad R, Huber I, Hormansdorfer S, Zols S, Eddicks M, Ritzmann M, Sing A. Corynebacterium species nasal carriage in pigs and their farmers in Bavaria, Germany: implications for public health. Vet Rec. 2014;175:248
7. Farfour E, Leto J, Barritault M, Barberis C, Meyer J, Dauphin B, Le Guern AS, Lefleche A, Badell E, Guiso N, Leclercq A, Le Monnier A, Lecuit M, Rodriguez-Nava V, Bergeron E, Raymond J, Vimont S, Bille E, Carbonnelle E, Guet-Revillet H, Lecuyer H, Beretti JL, Vay C, Berche P, Ferroni A, Nassif X, Join-Lambert O. Evaluation of the Andromas matrix-assisted laser desorption ionization-time of flight mass spectrometry system for identification of aerobically growing Gram-positive bacilli. J Clin Microbiol. 2012;50:2702-2707
8. Chevreux B, Wetter T, Suhai S. Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. Proceedings of the German Conference on Bioinformatics. 1999:45-56
9. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455-477
10. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75
Author contact
![]() Corresponding authors: Tel.: + 55 71 3283-8940. E-mail: luis.pachecobr (L. Pacheco) / rommelthiagocom (R.T. Ramos).
Corresponding authors: Tel.: + 55 71 3283-8940. E-mail: luis.pachecobr (L. Pacheco) / rommelthiagocom (R.T. Ramos).