ISSN: 1839-9940Journal of Genomics

J Genomics 2018; 6:41-52. doi:10.7150/jgen.24228 This volume Cite
Research Paper
De novo Assembly of the Burying Beetle Nicrophorus orbicollis (Coleoptera: Silphidae) Transcriptome Across Developmental Stages with Identification of Key Immune Transcripts
Harim I. Won, Thomas T. Schulze#, Emalie J. Clement, Gabrielle F. Watson, Sean M. Watson, Rosalie C. Warner, Elizabeth A. M. Ramler, Elias J. Witte, Mark A. Schoenbeck, Claudia M. Rauter, Paul H. Davis ![]()
Department of Biology, University of Nebraska at Omaha, Omaha, Nebraska.
#Current address: Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, Nebraska.
Abstract

Burying beetles (Nicrophorus spp.) are among the relatively few insects that provide parental care while not belonging to the eusocial insects such as ants or bees. This behavior incurs energy costs as evidenced by immune deficits and shorter life-spans in reproducing beetles. In the absence of an assembled transcriptome, relatively little is known concerning the molecular biology of these beetles. This work details the assembly and analysis of the Nicrophorus orbicollis transcriptome at multiple developmental stages. RNA-Seq reads were obtained by next-generation sequencing and the transcriptome was assembled using the Trinity assembler. Validation of the assembly was performed by functional characterization using Gene Ontology (GO), Eukaryotic Orthologous Groups (KOG), and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. Differential expression analysis highlights developmental stage-specific expression patterns, and immunity-related transcripts are discussed. The data presented provides a valuable molecular resource to aid further investigation into immunocompetence throughout this organism's sexual development.
Keywords: transcriptome, de novo assembly, immunity, parental care, burying beetle, Nicrophorus orbicollis
Introduction
The development of animals with complex life cycles is characterized by substantial changes in morphology, physiology, behavior, and often ecology [1,2]. Especially drastic changes occur during metamorphosis during the pupal stage, which occurs between larval and adult stages and which often includes a change in ecological niche [1,2]. The majority of insects (e.g., Lepidoptera, Hymenoptera, Diptera, Coleoptera [3]) undergo complete metamorphosis (i.e., are holometabolous) and have four differentiated life stages: egg, larva, pupa, and adult. These changes are also accompanied by changes in gene expression [4-7]. These previous studies have primarily focused on non-social or eusocial insects; little is known about changes in transcripts during development of subsocial insects, which form small groups consisting of mother and/or father and offspring with the parents providing parental care. Comparing the groups of transcripts which are differentially expressed in each developmental stage of subsocial insects can give insight into the molecular mechanisms of organismal development of subsocial organisms in general, including birds and mammals, and guide new directions of study.
Burying beetles (Coleoptera: Silphidae: Nicrophorus) are an excellent system for these kinds of studies. Notably, Nicrophorus spp. are known for providing extensive biparental care to their offspring [8-10], a feature which is uncommon even among the eusocial insects. Nicrophorus reproduction begins with an adult beetle using chemosensors to locate small vertebrate carrion, which serve as the primary larval food resource [8]. After locating the carrion, a physiological response is observed in the female beetle within a few hours: ovarian volume increases two- to three-fold [11]. To prepare the carrion, the female (or male if present) will bury the carrion and remove its exterior coat. The female then lays her eggs in the soil next to the buried carrion. Following oviposition, the eggs hatch and the first instar larvae emerge. Nicrophorus development features three instar stages, between which they shed their exoskeletons with altered morphology at each stage [9]. Both male and female parent will guard the brood against predators and feed begging larvae by regurgitating carrion into their mouths [12]. After three days, the larvae can feed autonomously, and the male parent leaves prior to the completion of larval development. The female parent, however, remains with the brood until it enters the pupal developmental stage [8]. Similar to most holometabolous insect species, N. orbicollis pupae are sessile and non-feeding; during this stage, larval insect structures are degraded and adult structures are developed [13]. Finally, the adult Nicrophorus emerge from pupae, mature sexually within the subsequent three weeks, and will then begin reproduction themselves upon discovery of a small vertebrate carrion.
For burying beetles, parental care incurs fitness costs in addition to the energetic costs of reproduction. This is illustrated by the substantially shorter life-spans of beetles which provide parental care in comparison to those beetles which only mate or produce eggs [14]. Female Nicrophorus beetles experience suppression of their personal immunity when providing parental care in favor of social immunity manifested by the production of antibacterial substances used to protect the progeny and breeding resources from infection [15-17]. These substances are produced as anal exudates that the parent coat on the carrion (on which they breed), demonstrate robust antimicrobial activity [18,19], and improve larval survival [20]. These immune and fitness costs of parental care are under evaluation among Nicrophorus species.
To date, much research on the molecular mechanisms of immunity during insect reproduction has focused on model organisms, such as fruit flies (Drosophila melanogaster), which do not provide parental care [21]. To begin to understand how parental care may induce fitness and immunity costs, basic knowledge of the molecular and physiological mechanisms underlying parental care are needed and can be derived, at least in part, from a knowledge of gene expression throughout development. The degree of an insect species' sociality may modify the expression of transcripts in the holometabolous insects (i.e., insects with metamorphosis). This current study compares transcriptomes of N. orbicollis, a burying beetle with obligate parental care, that were generated at various stages in the life cycle, including the egg, the larval stages, pupa, and sexually mature (though non-reproducing) adult. Developmental transcriptome profiles are available for non-social model systems with incomplete metamorphosis (i.e., hemimetabolous) such as the cricket Gryllus rubens [22] and acridid grasshopper Chorthippus biguttulus [23] and the subsocial, hemimetabolous European earwig Forficula auricularia [24]; life-spanning transcriptome analyses are also available for eusocial holometabolous systems, such as the bumblebee Bombus terrestris [4]. In contrast to these studies - and prior works examining Nicrophorus spp. - this work provides a characterization of the life-stage transcriptomes of an insect that is both subsocial and holometabolous.
Previous transcriptome analyses of Nicrophorus spp. had the goal of discovering the molecular bases of parental care behaviors. One study compared the transcriptomes of both male and female high- and low- provisioning parents of N. orbicollis and N. vespilloides; the comparison suggested that the variability of specific transcripts during the transition to parental care may condition the degree of provisioning [25]. The recruitment of genes mediating personal immunity for the function of social immunity was indicated by the identification of specific lysozyme transcripts up-regulated in breeding females [15], corresponding to the increased levels of lysozyme occurring in anal exudates employed for carrion maintenance. Further, it was demonstrated that the expression levels of different antimicrobial peptide-encoding transcripts were context-sensitive [26], changing according to sex and the occurrence of the carrion and offspring. Examination of the N. vespilloides gut tissue and microfloral transcriptomes [27] revealed spatial and temporal differentiation of transcripts encoding antimicrobial peptides and detoxifying enzymes, and detected microbial populations that might be beneficially transferred to the carrion.
Current molecular resources for N. orbicollis are just emerging and include a limited number of annotated nucleotide and protein sequences, as well as unassembled RNA-Seq reads of salivary glands of fasted and fed adult organisms (NCBI SRA: SRP092063). Notably, there are published transcriptomes for an organism in the same genus, Nicrophorus vespilloides [15,28]. However, it has been thoroughly documented that N. vespilloides provides facultative care, whereas N. orbicollis provides obligatory parental care. In facultative care, parental care merely enhances larval growth and is not necessary for survival. Conversely, in obligatory care, larvae do not survive in the absence of care [12,25,29-31]. We propose that this differentiation makes N. orbicollis an ideal organism in which to study the costs of Nicrophorus parental care on both fitness and immune function as a model for other organisms which feature obligate parental care.
In this work, the assembly and analysis of the N. orbicollis transcriptome are described. The aim of this study is to compare the transcriptomes of N. orbicollis eggs, all larval stages, pupae, and sexually mature, non-breeding females. Beetle samples obtained from an inbred laboratory colony in Omaha, Nebraska were sequenced by next-generation sequencing (NGS), followed by assembly of the de novo transcriptome with Trinity. Functional characterizations utilized to validate the transcriptome are included and discussed. The transcriptomic data presented in this work will aid further molecular studies in N. orbicollis and enable its development as a potential model organism for studying obligate parental care and its impact on reproducing adult organisms.
Methods
Insects: The beetles used in this study were from a stock colony of Nicrophorus orbicollis and descended from beetles that were caught at T.L. Davis Prairie, Nebraska, USA in 2008. Larvae from these field-caught beetles were reared by N. tomentosus foster parents which had been reared without parents to minimize internal and external parasites such as mites and nematodes. Each generation (i.e., every 9-11 weeks), 40 to 100 non-sibling N. orbicollis pairs were used as parents for the subsequent generation. Colony beetles were maintained individually in 15 × 29 × 11 cm plastic containers (Pioneer Plastics, Dixon, KY, USA) filled with 2 cm of moist peat at 20 ± 1°C in a 15:9 hr light:dark cycle. Twice a week, the beetles were fed canned cat food (Friskies® Nestlé Purina PetCare Company, St. Louis, MO, USA).
For this study, 6 pairs of N. orbicollis sisters were each mated to one of six brothers. The following day, the males were removed, and the females was placed into 15 × 29 × 11 cm plastic containers, filled with 2 cm moist peat. Each female received a Swiss-Webster mouse carcass (20-25 g, Charles River Laboratories, O'Fallon, MO, USA) that had been frozen shortly following death and had been thawed to 25 °C overnight. Breeding beetles were kept at 24 ± 1 °C in the dark to simulate underground conditions and were only removed for periodic inspection. Each of these females provided samples for one of the following life-history stages: approximately 10 individual eggs, 3 individual 1st instar larvae, 2 individual 2nd instar larvae, a single early 3rd instar larva, a single pupa, and a single sexually mature, non-reproducing adult female.
Tissue Collection and RNA Isolation: Beetles across the six developmental stages were sampled from the brood as development progressed from December 2016 to January 2017.
Beetle samples were rinsed in 70% ethanol to reduce potential ectoparasites and exterior bacteria, euthanized, and immediately stored at -20 °C in RNAlater® (Ambion Inc., Foster City, CA, USA) according to manufacturer instructions. Individual beetle samples were mechanically homogenized on ice and further homogenized with QIAshredder columns; an RNeasy Plus Mini Kit (Qiagen, Germantown, MD, USA) was then used to isolate whole organism RNA. Nucleic acid quantification was accomplished with a Thermo Scientific™ NanoDrop 2000c and RNA integrity was verified with a bleach denaturing agarose electrophoresis gel [32]. Prior to sequencing, purified RNA was stored at -80°C with minimal handling and freeze-thaw cycles.
Next-generation Sequencing: Sequencing was performed at the University of Nebraska Medical Center Genomics Core Facility. Initial quality control analysis to confirm the quality of the isolated RNA was done with the Fragment Analyzer™ Automated CE System (Advanced Analytical Technologies, Inc., Ankeny, IA, USA). An Illumina® TruSeq RNA Library Preparation Kit was used to produce the library prior to sequencing. An Illumina® NextSeq 500 Sequencing System was used to generate paired-end 151 bp reads. The 1st instar larvae, pupa, and non-breeding female adult RNA samples were selected for the representative transcriptome assembly, each occupying one lane on a NextSeq 500 flow cell for deep sequencing. The egg, 2nd instar larvae, and 3rd instar larva RNA samples shared the single remaining lane on the flow cell.
Assembly and Processing: Resultant sequencing reads were first evaluated using FastQC [33] to assess initial read quality. PRINSEQ [34] was then used to trim reads with a Phred quality score of <30 in a five base sliding window and a DUST threshold of 70. The trimmed paired-end reads were resynchronized [35] and de novo assembly was performed from these synchronized reads with the Trinity assembler (version 2.1.1) with the maximum memory set to 33 GB, number of CPUs set to 6, and with normalization of reads [36,37]. TransDecoder [36] was used to search this initial assembly for predicted coding sequences which were ≥50 amino acids long. The assembly was then BLASTed using default parameters against NCBI RefSeq metazoan protein sequences (retrieved October 2017); results with a bit score ≥50 were retained. Microbial sequences were also eliminated using BLAST [38]. Noncoding RNA was separated from the assembly by homology to the Rfam sequences of Drosophila melanogaster [39]. Finally, highly similar sequences were merged with CD-HIT [40] to produce the representative transcriptome. Both the final assembly and the representative transcriptome are available at http://www.davislab.net/nicrophorus/. The mapping rates of the paired-end reads of individual stage libraries to the representative transcriptome were obtained using Bowtie2 (version 2.1.0) [41] by alignment of paired-end reads to the representative transcriptome. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/ENA/GenBank under the accession GGAA00000000. The version described in this paper is the first version, GGAA01000000.
Transcript Coverage: A BLAST database was produced from the whole N. orbicollis assembly; all non-redundant Nicrophorus vespilloides and Tribolium castaneum protein sequences (retrieved from NCBI RefSeq October 2017) were then BLASTed against this database. The tBLASTn was limited to alignments with an e-value less than 10-5. The length of each alignment was compared to the length of the reference organism query sequence to acquire a percent coverage value. Counts of the unique sequences found in ranges of percent coverage were then obtained.
GO: The N. orbicollis translated protein assembly and all N. vespilloides, T. castaneum, and D. melanogaster protein sequences (retrieved from NCBI RefSeq October 2017) were BLASTed against the NCBI nr database (retrieved May 2017) GI-restricted to 11 of the 12 fully-annotated GO organisms (Drosophila melanogaster, Caenorhabditis elegans, Danio rerio, Gallus gallus, Homo sapiens, Rattus norvegicus, Mus musculus, Schizosaccharomyces pombe, Saccharomyces cerevisiae, Dictyostelium discoideum, and Arabidopsis thaliana). To avoid assigning GO-terms to any prokaryotic sequences which were not filtered, an Escherichia coli GI list was not included. The output BLAST results were analyzed by Blast2GO (version 2.8.0) using a local Blast2GO database (downloaded June 2017) [42] and GO terms were assigned for the proteins in each organism. Gene products of N. orbicollis and the reference organisms were classified by biological process, molecular function, and cellular component, and counts of these level 2 GO terms were obtained.
KOG: The translated N. orbicollis assembly and all N. vespilloides, T. castaneum, and D. melanogaster protein sequences (retrieved from NCBI RefSeq October 2017) were BLASTed against the NCBI KOG database (version 3.16) [43]. The RPS-BLAST was limited to alignments with an e-value less than 10-5. The top unique BLAST results showing the highest-scoring alignments for each respective query were identified and the respective Conserved Domains Database (CDD) ID was retained. CDD IDs were matched to KOG IDs and counts of individual KOG groups in each organism were obtained.
KEGG: The representative N. orbicollis assembly and all N. vespilloides, T. castaneum, and D. melanogaster mRNA sequences (retrieved from NCBI RefSeq October 2017) were uploaded to the KEGG Automatic Annotation Server (KAAS) and processed by BLAST comparisons against a database of KEGG genes [44]. Ortholog assignments were returned and counts were obtained.
Genes of Interest: Genes known in the literature to be involved in both personal and social immunity of insects, particularly N. vespilloides, were selected and their respective homologs in N. orbicollis were identified. The N. orbicollis annotation file, described below, was utilized to match the homologs for each selected N. vespilloides immune gene.
Differential Expression: Differential expression in the developmental stages was examined in the egg, 1st instar larva, 2nd instar larva, 3rd instar larva, pupa, and fully-developed, non-breeding female adult transcriptomes. Transcript abundance in each respective sample was estimated by aligning raw reads from each sample separately to the code determining sequence (CDS) representative transcriptome assembly utilizing the short read aligner, Bowtie2 [41]. Estimates of abundance were then generated using the Trinity script, abundance_estimates_to_matrix [37], with RNA-Seq by Expectation Maximization (RSEM) [45]; of the different absolute and normalized transcript count formats given, normalized expression values were selected in the Transcripts Per Kilobase Million (TPM) format. TPM counts were then log2 transformed. Transcript annotation was accomplished by homology to SwissProt database proteins (retrieved November 2017) and all N. vespilloides protein sequences retrieved from the NCBI nr database (retrieved May 2017). FASTA files for containing putative annotations for the representative transcriptome are available for download at http://www.davislab.net/nicrophorus.
Results and Discussion
Transcriptome Characteristics
Raw NGS reads have been deposited at NCBI SRA under accession SRP124908. Assemblies include a representative assembly consisting of 21,409 unique transcripts, a complete nucleotide assembly, and a translated protein assembly. The representative transcriptome was assembled de novo with RNA samples from three distinct life stages (see Methods) using the Trinity assembler. Illumina® paired-end reads, resulting in 66.7 G bases of output (Table 1), were utilized to generate the assembly. General characteristics of the representative transcriptome and overall paired-end reads mapping rates of individual libraries to the representative transcriptome are reported in Table 2.
Assessing Full-length Transcript Coverage
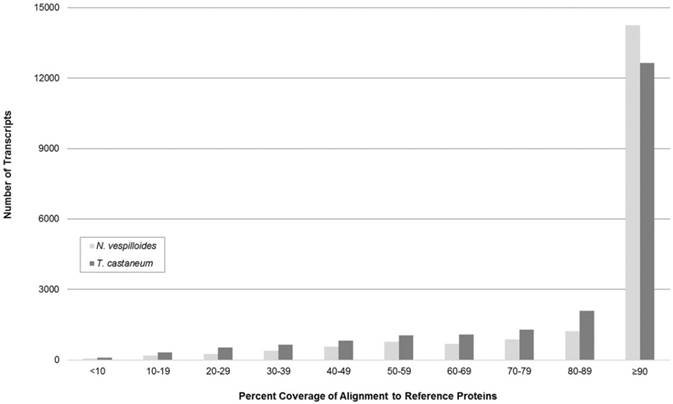
To evaluate the completeness of the transcripts comprising the assembled representative transcriptome, coverage histograms were generated from alignments with non-redundant protein sequences from two model organisms: N. vespilloides and T. castaneum. The availability of publicly accessible transcriptomes determined which model organisms were selected for comparison. tBLASTn alignment length coverage that exceeded 90% of the reference organism's protein length was seen in 72.8% of N. orbicollis transcripts after alignment with N. vespilloides proteins and 55.9% with T. castaneum proteins (Figure 1). Considering genetic variations among the selected organisms, the histogram suggests that the produced representative transcriptome contains a high abundance of full-length transcripts.
Transcriptome Sequencing Details. RNA yields ranged from 3.7 to 64.0 µg of purified RNA for beetles across developmental life stages. Sequencing was performed on an Illumina® NextSeq 500. Overall paired-end reads mapping rates of individual sample libraries are included. The representative transcriptome has been filtered for ribosomal RNA, noncoding RNA, microbial and plant contaminating RNA, and mitochondrial sequences; therefore, any reads in the individual libraries mapping to these filtered sequences would not map to the representative transcriptome. bp: base pair, GC: G-C nucleotide ratio
| Tissue | Reads | Bases (bp) | GC Content | Mapping Rate |
|---|---|---|---|---|
| Eggs | 39,722,314 | 5,798,921,254 | 46% | 90.5% |
| First instar larvae | 127,028,854 | 18,509,700,969 | 45% | 89.3% |
| Second instar larvae | 35,479,130 | 5,167,790,213 | 46% | 85.2% |
| Third instar larva | 39,298,572 | 5,720,171,102 | 46% | 87.0% |
| Pupa | 111,438,838 | 16,278,597,449 | 45% | 85.0% |
| Non-breeding female adult | 103,768,508 | 15,199,597,722 | 42% | 79.4% |
| Total | 456,736,216 | 66,674,778,709 | 45% | 86.1% |
Representative Transcriptome Characteristics. The representative Nicrophorus orbicollis transcriptome assembly was analyzed and the above characteristics were determined. In comparison, 65,280 transcripts were assembled from the initial raw output.
| Total Transcripts | Mean Length (bp) | Median Length (bp) | N50 (bp) | GC Content |
|---|---|---|---|---|
| 21,409 | 1,762.31 | 1,193 | 2,856 | 42.1% |
Coverage Length of Nicrophorus orbicollis Transcripts. Coverage of Nicrophorus vespilloides and Tribolium castaneum translated nucleotide transcripts by N. orbicollis assembled sequences is shown. The N. vespilloides (count = 19,579) and T. castaneum (count = 22,630) proteomes (retrieved from NCBI RefSeq) were compared to the representative N. orbicollis assembly with tBLASTn. The majority of N. orbicollis transcripts showed greater than 90% coverage length of reference organism protein sequences in both N. vespilloides (72.8%) and T. castaneum (55.9%), suggesting a high-degree of full-length transcripts in the representative assembly.
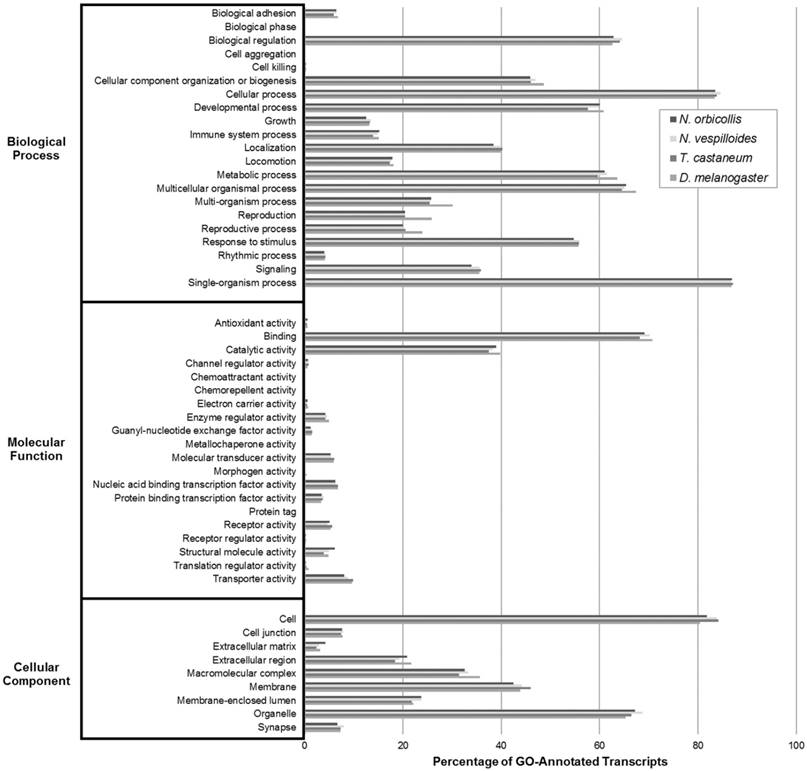
Gene Ontology (GO) Transcriptomic Analysis. GO analysis was performed on Nicrophorus orbicollis predicted proteins and the Nicrophorus vespilloides, Tribolium castaneum, and Drosophila melanogaster proteomes (retrieved from NCBI RefSeq) to functionally categorize the translated assembly with level 2 GO terms and assess its completeness. The GO term distributions of the organisms closely match each other, suggesting that the assembled N. orbicollis transcriptome is fairly complete.
Transcriptome Functional Characterization
Three analyses grouping transcripts into broad putative functional categories were utilized to annotate and assess the N. orbicollis transcriptome assembly. These were Gene Ontology (GO), Eukaryotic Orthologous Groups (KOG), and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. These grouping tools were used to examine the general functions of the assembled transcripts and to assess the quality and completeness of the assembly in direct comparison to reference organisms with well-annotated transcriptomes, N. vespilloides, T. castaneum, and D. melanogaster. The GO analysis compared the N. orbicollis putative proteome against those of the model GO organisms. For the level 2 GO terms, Biological Process, Molecular Function, and Cellular Component, the functional groups under which the most transcripts were annotated were single-organism process (87.0%), binding (69.2%), and organelle (67.2%), respectively. The degree of similarity in the GO groupings among N. orbicollis and the reference organisms detailed in Figure 2 indicate that these insects share a functional distribution of their respective gene products.
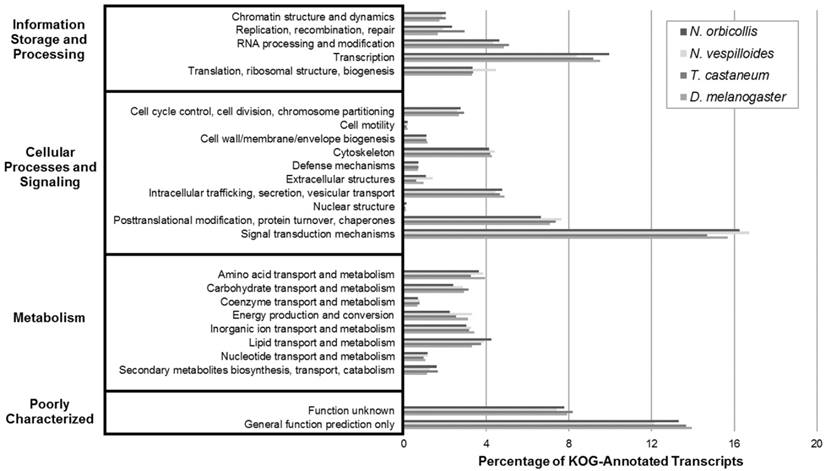
Next, the KOG analysis queried the N. orbicollis translated protein assembly against those of a set of seven eukaryotic model organisms, annotated proteins according to their function, and categorized proteins into clusters of eukaryotic orthologous groups. Under Information Storage and Processing, Cellular Processes and Signaling, and Metabolism, the orthologous groups which the most transcripts were assigned to were transcription (10.0%), signal transduction mechanisms (16.3%), and lipid transport and metabolism (4.2%), respectively. The KOG analysis of N. orbicollis resulted in classifying 79% of transcripts into KOG groups, and the comparable distributions of the KOG groupings also suggest a shared arrangement of gene products (Figure 3).
Eukaryotic Orthologous Groups (KOG) Transcriptomic Analysis. KOG analysis was performed on the Nicrophorus orbicollis predicted proteins and the Nicrophorus vespilloides, Tribolium castaneum, and Drosophila melanogaster proteomes (retrieved from NCBI RefSeq) to assess function and completeness of the transcriptome. KOG terms were assigned to each proteome; similar distributions are shown for each organism, supporting the completeness of the N. orbicollis transcriptome.
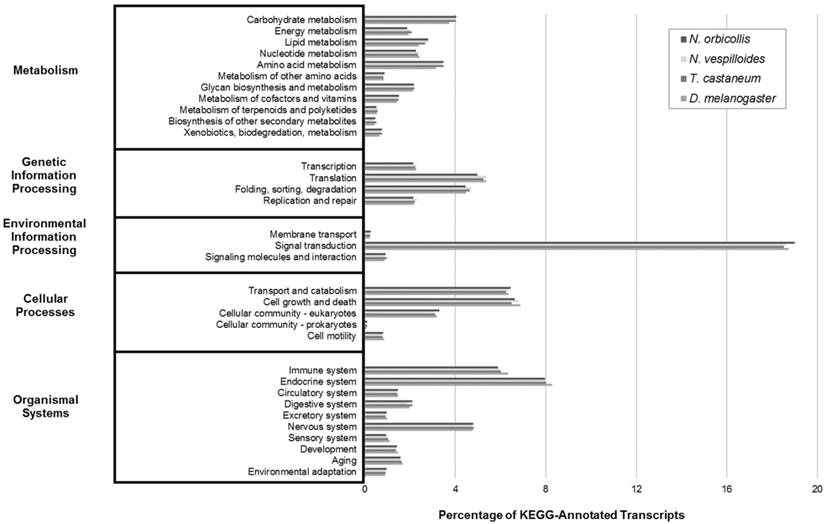
Finally, KEGG analysis was performed on the N. orbicollis predicted protein products to identify biologically relevant functional pathways through BLAST comparison against the KEGG databases of 24 completely sequenced genomes and their respective functional pathway categorizations. For Metabolism, Genetic Information Processing, Environmental Information Processing, Cellular Processes, and Organismal Systems, the functional pathways which the most transcripts were identified under were carbohydrate metabolism (4.0%), translation (5.0%), signal transduction (19.0%), cell growth and death (6.6%), and endocrine system (8.0%), respectively. Among N. orbicollis and the reference organisms, this analysis showed a consistent distribution of KEGG group numbers (Figure 4). Overall, each functional categorization algorithm showed close similarity among N. orbicollis and the selected reference organisms, indicating evidence in support of the completeness and consistency of the assembled transcriptome.
Identification of Key Immune Transcripts
The transcriptome outlined in this work provides a curated molecular resource which can be used in the investigation of care-related immunity losses in N. orbicollis. These beetles present a candidate model organism for studying these decreases due to the relative ease with which they are bred in the laboratory and their much quicker generation time [46] compared to that of mammals which provide parental care. Insect immunology is a continually growing field and various insects have been used as models to study infectious disease, the epigenetic basis of disease, inflammation and cancer, and adaptive immunity [47-49]. For example, wax moths (Galleria mellonella) have been used to study Legionella pneumophila, the etiologic agent of Legionnaires' disease. The larval stage of this moth has served as a model of infection of L. pneumophila as most murine strains are not permissive to infection with this human pathogen; an additional advantage of utilizing this species as an infection model is the throughput with which pathogen mutants can be screened to determine key virulence factors [50]. The increasing availability of transcriptomic resources continues to advance the field of insect immunology [15,51-54], providing a wealth of new model organisms in which to investigate immune function.
Immune-related Transcripts. Nicrophorus orbicollis genes linked to immune function were identified by homology to previously annotated proteins in the SwissProt database and Nicrophorus vespilloides NCBI RefSeq proteins for the development of N. orbicollis as a model organism for personal immunity deficits resulting from obligate parental care. Of the various genes with identified roles in immunity and parental care, a subset of annotated N. orbicollis homologs in the representative transcriptome were selected and are shown. Protein isoforms were distinguished by the annotation of the BLAST hit with the highest bit score in each respective transcript.
| Immune Function | Gene Name | Gene Symbol | Transcriptomic ID | Transcript Length (bp) |
|---|---|---|---|---|
| Antimicrobial Peptides | Cecropin C | CEC1 | NORBI01_22887 | 198 |
| Defensin | DEF | NORBI01_10621 | 327 | |
| Social Immunity | Lysozyme | LYS | NORBI01_5695 | 447 |
| Personal Immunity | Phenoloxidase 2 | PO2 | NORBI01_46051 | 2055 |
| Immunity Transfer | Vitellogenin 5 | VTG5 | NORBI01_13880 | 4230 |
Kyoto Encyclopedia of Genes and Genomes (KEGG) Transcriptomic Analysis. KEGG analysis was performed to describe transcript functions and to evaluate for transcriptome completeness. The Nicrophorus orbicollis representative assembly and the Nicrophorus vespilloides, Tribolium castaneum, and Drosophila melanogaster transcriptomes (retrieved from NCBI RefSeq) were functionally characterized into KEGG pathways. The percent distributions show similar proportions among the compared organisms, indicating a complete N. orbicollis transcriptome.
Previous studies have investigated the interface of personal and social immunity in insects and the trade-offs which are involved. Genes related to the effect parental care has on these functions of immunity are outlined in the literature and a selection of key genes from among those presented in these studies was tabulated and is illustrated in Table 3. The antimicrobial peptides cecropin C and defensin were identified as compounds present in oral and anal secretions of N. vespilloides, produced by breeding beetles as a parental investment in social immunity, increasing survival of larvae [26]. Defensin derived from T. castaneum has been shown to have efficacy against clinical isolates of multidrug-resistant Staphylococcus aureus [55], demonstrating the robustness of this feature of insect innate immunity. Lysozymes, additional innate immunity effectors which hydrolyze the structural polysaccharides of bacterial cell walls, comprise an important aspect of personal immunity [16,56] Several lysozymes have been identified in N. vespilloides with lysozyme 6 being investigated as a potential effector of social immunity recruited originally from personal immunity [15].
Phenoloxidase (PO) activity is a well-characterized component of the invertebrate personal immune response [57,58]. When the female burying beetles mate on a carcass, PO activity is suppressed in the hemolymph and induces an immune deficit likely in response to parental care activities. This effect is reversible in breeding N. vespilloides beetles, as they can increase PO activity in response to damage, but not to pre-reproduction expression levels [59].
Vitellogenin is as an egg yolk lipoprotein precursor which is essential for oocyte development; however, the literature indicates that it is also downregulated during active parental care in both parents, suggesting an additional associated role for vitellogenin in burying beetles [60]. In the eusocial honey bee, Apis mellifera, offspring immune priming is accomplished in the absence of antibodies by the transfer of immune elicitors directly into developing oocytes. Interestingly, it appears that vitellogenin binds to bacteria and pattern-associated molecular patterns (e.g., lipopolysaccharide, zymosan, and peptidoglycan) and is the protein carrier of these immune elicitors to insect eggs [61]. The described genes address various areas of investigation in Nicrophorus immunity and invite further investigation.
Differential Expression Analysis
A set of unique N. orbicollis transcripts which were differentially upregulated across developmental stages is described in Table 4. Transcripts which were either highly-expressed in one stage or highly-expressed across several stages were selected to highlight developmental progression. The expression levels of each stage were log2 transformed from raw TPM expression values. Substantially differentially expressed genes are discussed here.
Differentially Upregulated Transcripts in Developmental Stages. Nicrophorus orbicollis expression from six developmental stages was examined to assess differential transcript expression. Transcripts were identified by homology to previously annotated proteins in the SwissProt and NCBI RefSeq databases. Only the most differentially expressed isoform of each protein is shown.
| Transcriptomic ID | Log2 TPM | Protein Name |
|---|---|---|
| Egg stage differentially upregulated transcripts | ||
| NORBI01_35964 | 8.01 | hornerin |
| NORBI01_4444 | 6.86 | ferritin subunit |
| 1st Instar Larva stage differentially upregulated transcripts | ||
| NORBI01_35964 | 10.03 | hornerin |
| NORBI01_35941 | 9.98 | larval cuticle protein LCP-30 |
| NORBI01_13239 | 5.24 | semaphorin-5B |
| 2nd Instar Larva stage differentially upregulated transcripts | ||
| NORBI01_35964 | 10.96 | hornerin |
| NORBI01_35941 | 11.20 | larval cuticle protein LCP-30 |
| NORBI01_13239 | 9.90 | semaphorin-5B |
| 3rd Instar Larva stage differentially upregulated transcripts | ||
| NORBI01_35964 | 11.53 | hornerin |
| NORBI01_35941 | 11.74 | larval cuticle protein LCP-30 |
| NORBI01_13239 | 10.83 | semaphorin-5B |
| Pupa stage differentially upregulated transcripts | ||
| NORBI01_34488 | 10.76 | pupal cuticle protein |
| NORBI01_57190 | 12.44 | pupal cuticle protein 36 |
| NORBI01_10506 | 9.18 | pupal cuticle protein C1B |
| Adult stage differentially upregulated transcripts | ||
| NORBI01_63782 | 11.82 | vitellogenin |
| NORBI01_10621 | 11.47 | defensin |
Ferritin, with a log2 TPM value of 6.86 in the N. orbicollis egg sample, is a component of iron metabolism and is universal to almost all living organisms, including insects. In contrast to mammalian ferritins, which are cytoplasmic and function in iron storage, insect ferritins are typically associated with the vacuolar system and serve as iron transporters. It has been found that for hematophagous insects, 77% of absorbed non-heme iron is delivered to the ovaries and eggs in molecular association with ferritin [62]. While the genus Nicrophorus is not hematophagous, it has been shown that non-heme iron accumulates with age in rats [63]; with a breeding female beetle's consumption of carrion, and therefore non-heme iron, it is reasonable to expect that ferritin associates with this excess iron and navigates to the eggs. As the hemolymphatic system develops within the eggs, there is a need to export the accumulated iron to developing cells for use in oxidative metabolism [64]; we expect and observe upregulation of this transporter protein in the life stage with excess iron as a result of maternal transfer.
Upregulated in the egg and all larval stages, hornerin is a structural protein which includes a collagen triple helix repeat [65], as well as a chitin-binding Peritrophin-A domain [66]. The function of this protein is not well characterized in beetles, but it can be inferred from the conserved functional domains [67] and differential expression results that this protein is involved in forming connective tissue structure and in binding chitin found in the peritrophic matrix of the egg and larval stages of Nicrophorus. Through the egg and each instar stage, a hornerin log2 TPM value range of 10.03 to 11.53 is observed. Notably, hornerin is not upregulated in the pupal stage; as the organism undergoes metamorphosis, its structural needs adjust. Particularly since pupation is a non-feeding developmental stage, it is conceivable that N. orbicollis pupae do not require this protein which binds a structure so involved in the digestive biology of the insect.
A similar stepwise increase in larval cuticle protein LCP-30 expression from 9.98 to 11.74 log2 TPM is detected through the three larval instar stages. This gene product is another structural protein; the larval cuticle protein isoforms and chitin form the structural constituents of the larval cuticle. The R and R consensus motif found within many insect cuticle proteins have been shown to bind chitin, but have no sequence homology to the chitin-binding domain of peritrophic membrane proteins like hornerin [68]. In the pupal stage, three pupal cuticle protein isoforms are highlighted with expression values ranging from 9.18 to 12.44. The expression profiles of the selected larval and pupal cuticular proteins show developmental stage-appropriate upregulation, as would be expected.
Semaphorin 5b, a plexin receptor family semaphorin, is upregulated progressively through the larval stages (5.24 to 10.83 log2 TPM expression values) and this family of proteins has been shown to be important for neural system development by functioning as axonal growth cone guidance molecules [69]. A previous study in D. melanogaster found semaphorin 2 to inhibit certain synaptic terminal arbors during motoneuron outgrowth and synapse formation [70]. Additional studies have shown that these proteins are critical for functional neuronal connection assembly [71] and that some can also play roles in immune regulation and signaling - particularly among the plexin-family semaphorins [72,73]. Our data could suggest that a critical stage of Nicrophorus neural development and/or immunoregulation occurs during developmental progression through the larval stages.
Finally, vitellogenin is observed to have an expression value of 11.82 in the sexually mature, non-breeding adult female. This protein is described above and its function as an egg yolk lipoprotein precursor and its potential involvement in parental immunity [60,61] both highlight vitellogenin as a protein expected to be differentially expressed in this developmental stage. Defensin is also seen to be upregulated (11.47 log2 TPM) in this stage. This antimicrobial peptide is also discussed above, and its secretion by adults as a component of parental care and social immunity [26,55] naturally associates this protein's expression with the adult stage.
Conclusion
By functional characterization, we have been able to validate the quality and completeness of our N. orbicollis transcriptome assembly. The availability of this transcriptome presents a valuable molecular resource for the investigation of this species as a model to study the immunological deficits which directly result from providing parental care. The species in this genus present a niche of insects which provide biparental care - a niche which is further narrowed by the obligate parental care observed in N. orbicollis.
Abbreviations
NGS: next-generation sequencing; GO: gene ontology; KOG: eukaryotic orthologous groups; CDD: Conserved Domains Database; KEGG: Kyoto encyclopedia of genes and genomes; KAAS: KEGG Automatic Annotation Server; SRA: Sequence Reads Archive; RSEM: RNA-Seq by Expectation Maximization; TPM: Transcripts Per Kilobase Million.
Acknowledgements
This work was supported by NIH grant P20GM103427. Additional financial support was provided by a University of Nebraska at Omaha Fund for Undergraduate Scholarly Experiences (FUSE) grant. HIW was also partially supported by the Barry Goldwater Scholarship. The University of Nebraska Medical Center Genomics Core Facility provided RNA sequencing and receives partial support from the Nebraska Research Network in Functional Genomics, The Molecular Biology of Neurosensory Systems CoBRE P30GM110768, The Fred & Pamela Buffett Cancer Center P30CA036727, The Center for Root and Rhizobiome Innovation (CRRI) 36-5150-2085-20, and the Nebraska Research Initiative. This work was also completed utilizing the Holland Computing Center of the University of Nebraska which receives support from the Nebraska Research Initiative. This publication's contents are the sole responsibility of the authors and do not necessarily represent the official views of the funding agencies.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wilbur HM. Complex Life Cycles. Annu Rev Ecol Syst. 1980;11:67-93 doi:10.1146/annurev.es.11.110180.000435
2. Minelli A, Fusco G. Developmental plasticity and the evolution of animal complex life cycles. Philos Trans R Soc B Biol Sci. 2010;365:631-640 doi:10.1098/rstb.2009.0268
3. Mayhew PJ. A tale of two analyses: estimating the consequences of shifts in hexapod diversification. Biol J Linn Soc. 2003;80:23-36 doi:10.1046/j.1095-8312.2003.00217.x
4. Colgan TJ, Carolan JC, Bridgett SJ, Sumner S, Blaxter ML, Brown MJ. Polyphenism in social insects: insights from a transcriptome-wide analysis of gene expression in the life stages of the key pollinator, Bombus terrestris. BMC Genomics. 2011;12:623. doi:10.1186/1471-2164-12-623
5. Harker BW, Behura SK, deBruyn BS, Lovin DD, Mori A, Romero-Severson J. et al. Stage-specific transcription during development of Aedes aegypti. BMC Dev Biol. 2013;13:29. doi:10.1186/1471-213X-13-29
6. Sayadi A, Immonen E, Bayram H, Arnqvist G. The De Novo Transcriptome and Its Functional Annotation in the Seed Beetle Callosobruchus maculatus. PloS One. 2016;11:e0158565. doi:10.1371/journal.pone.0158565
7. Zhang T, He K, Wang Z. Transcriptome Comparison Analysis of Ostrinia furnacalis in Four Developmental Stages. Sci Rep. 2016;6:35008. doi:10.1038/srep35008
8. Scott MP. The ecology and behavior of burying beetles. Annu Rev Entomol. 1998;43:595-618 doi:10.1146/annurev.ento.43.1.595
9. Pukowski E. Ökologische Untersuchungen an Necrophorus F. Z Für Morphol Ökol Tiere. 1933;27:518-586 doi:10.1007/BF00403155
10. Eggert AK, Muller JK. Biparental care and social evolution in burying beetles: lessons from the larder. In: (ed.) Choe JC, Crespi BJ. The Evolution of Social Behavior in Insects and Arachnids, Cambridge: Cambridge Univ. Press. 1997:216-236 Available: http://agris.fao.org/agris-search/search.do?recordID=GB1997039354
11. Trumbo ST. Juvenile hormone-mediated reproduction in burying beetles: From behavior to physiology. Arch Insect Biochem Physiol. 1997;35:479-490 doi:10.1002/(SICI)1520-6327(1997)35:4<479::AID-ARCH9>3.0.CO;2-M
12. Eggert A-K, Reinking M, Müller JK. Parental care improves offspring survival and growth in burying beetles. Anim Behav. 1998;55:97-107 doi:10.1006/anbe.1997.0588
13. Borror DJ, Johnson NF, Triplehorn CA. An Introduction to the Study of Insects. Saunders College Pub. 1989
14. Trumbo S, Rauter C. Juvenile hormone, metabolic rate, body mass and longevity costs in parenting burying beetles. Anim Behav. 2014;92:203-211 doi:10.1016/j.anbehav.2014.04.004
15. Palmer WJ, Duarte A, Schrader M, Day JP, Kilner R, Jiggins FM. A gene associated with social immunity in the burying beetle Nicrophorus vespilloides. Proc R Soc B. 2016;283:20152733. doi:10.1098/rspb.2015.2733
16. Hultmark D. Insect lysozymes. EXS. 1996;75:87-102
17. Daffre S, Kylsten P, Samakovlis C, Hultmark D. The lysozyme locus in Drosophila melanogaster: an expanded gene family adapted for expression in the digestive tract. Mol Gen Genet MGG. 1994;242:152-162
18. Cotter SC, Littlefair JE, Grantham PJ, Kilner RM. A direct physiological trade-off between personal and social immunity. J Anim Ecol. 2013;82:846-853 doi:10.1111/1365-2656.12047
19. Cotter SC, Topham E, Price AJP, Kilner RM. Fitness costs associated with mounting a social immune response. Ecol Lett. 2010;13:1114-1123 doi:10.1111/j.1461-0248.2010.01500.x
20. Arce AN, Johnston PR, Smiseth PT, Rozen DE. Mechanisms and fitness effects of antibacterial defences in a carrion beetle. J Evol Biol. 2012;25:930-937 doi:10.1111/j.1420-9101.2012.02486.x
21. Viljakainen L. Evolutionary genetics of insect innate immunity. Brief Funct Genomics. 2015;14:407-412 doi:10.1093/bfgp/elv002
22. Berdan EL, Blankers T, Waurick I, Mazzoni CJ, Mayer F. A genes eye view of ontogeny: de novo assembly and profiling of the Gryllus rubens transcriptome. Mol Ecol Resour. 2016;16:1478-1490 doi:10.1111/1755-0998.12530
23. Berdan EL, Finck J, Johnston PR, Waurick I, Mazzoni CJ, Mayer F. Transcriptome profiling of ontogeny in the acridid grasshopper Chorthippus biguttulus. PLOS ONE. 2017;12:e0177367. doi:10.1371/journal.pone.0177367
24. Roulin AC, Wu M, Pichon S, Arbore R, Kühn-Bühlmann S, Kölliker M. et al. De Novo Transcriptome Hybrid Assembly and Validation in the European Earwig (Dermaptera, Forficula auricularia). PLOS ONE. 2014;9:e94098. doi:10.1371/journal.pone.0094098
25. Benowitz KM, McKinney EC, Moore AJ. Difference in parenting in two species of burying beetle, Nicrophorus orbicollis and Nicrophorus vespilloides. J Ethol. 2016;34:315-319
26. Jacobs CGC, Steiger S, Heckel DG, Wielsch N, Vilcinskas A, Vogel H. Sex, offspring and carcass determine antimicrobial peptide expression in the burying beetle. Sci Rep. 2016:6 doi:10.1038/srep25409
27. Vogel H, Shukla SP, Engl T, Weiss B, Fischer R, Steiger S. et al. The digestive and defensive basis of carcass utilization by the burying beetle and its microbiota. Nat Commun. 2017;8:ncomms15186. doi:10.1038/ncomms15186
28. Parker DJ, Cunningham CB, Walling CA, Stamper CE, Head ML, Roy-Zokan EM. et al. Transcriptomes of parents identify parenting strategies and sexual conflict in a subsocial beetle. Nat Commun. 2015;6:8449. doi:10.1038/ncomms9449
29. Capodeanu-Nägler A, Keppner E, Vogel H, Ayasse M, Eggert A, Sakaluk S. et al. From facultative to obligatory parental care: Interspecific variation in offspring dependency on post-hatching care in burying beetles. Sci Rep. 2016:6 doi:10.1038/srep29323
30. Schrader M, Jarrett BJM, Kilner RM. Using Experimental Evolution to Study Adaptations for Life within the Family. Am Nat. 2015;185:610-619 doi:10.1086/680500
31. Trumbo ST. Monogamy to communal breeding: exploitation of a broad resource base by burying beetles (Nicrophorus). Ecol Entomol. 1992;17:289-298 doi:10.1111/j.1365-2311.1992.tb01060.x
32. Aranda PS, LaJoie DM, Jorcyk CL. Bleach gel: a simple agarose gel for analyzing RNA quality. Electrophoresis. 2012;33:366-369 doi:10.1002/elps.201100335
33. Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010 [cited 24 Oct 2017]. Available: https://. www.bioinformatics.babraham.ac.uk/projects/fastqc/
34. Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinforma Oxf Engl. 2011;27:863-864 doi:10.1093/bioinformatics/btr026
35. Normandeau E. fastqCombinePairedEnd.py [Internet]. 2017. Available: https://github.com/enormandeau/Scripts.
36. Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494-1512 doi:10.1038/nprot.2013.084
37. G Grabherr M, Haas B, Yassour M, Z Levin J, Thompson D, Amit I. et al. Full-Length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644-52 doi:10.1038/nbt.1883
38. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K. et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi:10.1186/1471-2105-10-421
39. Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y. et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016;44:D203-208 doi:10.1093/nar/gkv1252
40. Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinforma Oxf Engl. 2012;28:3150-3152 doi:10.1093/bioinformatics/bts565
41. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357-359 doi:10.1038/nmeth.1923
42. Gene Ontology Consortium. The Gene Ontology project in 2008. Nucleic Acids Res. 2008;36:D440-D444 doi:10.1093/nar/gkm883
43. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi:10.1186/1471-2105-4-41
44. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27-30
45. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi:10.1186/1471-2105-12-323
46. Rauter CM, Moore AJ. Evolutionary importance of parental care performance, food resources, and direct and indirect genetic effects in a burying beetle. J Evol Biol. 2002;15:407-417 doi:10.1046/j.1420-9101.2002.00412.x
47. Mukherjee K, Twyman RM, Vilcinskas A. Insects as models to study the epigenetic basis of disease. Prog Biophys Mol Biol. 2015;118:69-78 doi:10.1016/j.pbiomolbio.2015.02.009
48. Apidianakis Y, Ferrandon D. Model organisms in inflammation and cancer. Frontiers E-books. 2014
49. Cooper D, Eleftherianos I. Memory and Specificity in the Insect Immune System: Current Perspectives and Future Challenges. Front Immunol. 2017:8 doi:10.3389/fimmu.2017.00539
50. Harding CR, Schroeder GN, Collins JW, Frankel G. Use of Galleria mellonella as a Model Organism to Study Legionella pneumophila Infection. J Vis Exp JoVE. 2013 doi:10.3791/50964
51. Bao Y-Y, Qu L-Y, Zhao D, Chen L-B, Jin H-Y, Xu L-M. et al. The genome- and transcriptome-wide analysis of innate immunity in the brown planthopper, Nilaparvata lugens. BMC Genomics. 2013;14:160. doi:10.1186/1471-2164-14-160
52. Zhang W, Chen J, Keyhani NO, Zhang Z, Li S, Xia Y. Comparative transcriptomic analysis of immune responses of the migratory locust, Locusta migratoria, to challenge by the fungal insect pathogen, Metarhizium acridum. BMC Genomics. 2015:16 doi:10.1186/s12864-015-2089-9
53. Xia X, Yu L, Xue M, Yu X, Vasseur L, Gurr GM. et al. Genome-wide characterization and expression profiling of immune genes in the diamondback moth, Plutella xylostella (L.). Sci Rep. 2015;5:srep09877. doi:10.1038/srep09877
54. Zhu J-Y, Yang P, Zhang Z, Wu G-X, Yang B. Transcriptomic Immune Response of Tenebrio molitor Pupae to Parasitization by Scleroderma guani. PLOS ONE. 2013;8:e54411. doi:10.1371/journal.pone.0054411
55. Rajamuthiah R, Jayamani E, Conery AL, Fuchs BB, Kim W, Johnston T. et al. A Defensin from the Model Beetle Tribolium castaneum Acts Synergistically with Telavancin and Daptomycin against Multidrug Resistant Staphylococcus aureus. PLoS ONE. 2015:10 doi:10.1371/journal.pone.0128576
56. Hoffmann JA. Innate immunity of insects. Curr Opin Immunol. 1995;7:4-10
57. Rodriguez-Andres J, Rani S, Varjak M, Chase-Topping ME, Beck MH, Ferguson MC. et al. Phenoloxidase Activity Acts as a Mosquito Innate Immune Response against Infection with Semliki Forest Virus. PLOS Pathog. 2012;8:e1002977. doi:10.1371/journal.ppat.1002977
58. Lu A, Zhang Q, Zhang J, Yang B, Wu K, Xie W. et al. Insect prophenoloxidase: the view beyond immunity. Front Physiol. 2014:5 doi:10.3389/fphys.2014.00252
59. Reavey CE, Warnock ND, Vogel H, Cotter SC. Trade-offs between personal immunity and reproduction in the burying beetle, Nicrophorus vespilloides. Behav Ecol. 2014;25:415-423 doi:10.1093/beheco/art127
60. Roy-Zokan EM, Cunningham CB, Hebb LE, McKinney EC, Moore AJ. Vitellogenin and vitellogenin receptor gene expression is associated with male and female parenting in a subsocial insect. Proc Biol Sci. 2015;282:20150787. doi:10.1098/rspb.2015.0787
61. Salmela H, Amdam GV, Freitak D. Transfer of Immunity from Mother to Offspring Is Mediated via Egg-Yolk Protein Vitellogenin. PLOS Pathog. 2015;11:e1005015. doi:10.1371/journal.ppat.1005015
62. Pham DQD, Winzerling JJ. Insect Ferritins: typical or atypical?. Biochim Biophys Acta. 2010;1800:824-833 doi:10.1016/j.bbagen.2010.03.004
63. Seo AY, Xu J, Servais S, Hofer T, Marzetti E, Wohlgemuth SE. et al. Mitochondrial iron accumulation with age and functional consequences. Aging Cell. 2008;7:706-716
64. Nichol H, Law JH, Winzerling JJ. Iron metabolism in insects. Annu Rev Entomol. 2002;47:535-559 doi:10.1146/annurev.ento.47.091201.145237
65. Mayne R, Brewton RG. New members of the collagen superfamily. Curr Opin Cell Biol. 1993;5:883-890
66. Shen Z, Jacobs-Lorena M. A type I peritrophic matrix protein from the malaria vector Anopheles gambiae binds to chitin. Cloning, expression, and characterization. J Biol Chem. 1998;273:17665-17670
67. Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S. et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45:D200-D203 doi:10.1093/nar/gkw1129
68. Rebers JE, Willis JH. A conserved domain in arthropod cuticular proteins binds chitin. Insect Biochem Mol Biol. 2001;31:1083-1093
69. Yazdani U, Terman JR. The semaphorins. Genome Biol. 2006;7:211. doi:10.1186/gb-2006-7-3-211
70. Matthes DJ, Sink H, Kolodkin AL, Goodman CS. Semaphorin II can function as a selective inhibitor of specific synaptic arborizations. Cell. 1995;81:631-639 doi:10.1016/0092-8674(95)90084-5
71. Pasterkamp RJ. Getting neural circuits into shape with semaphorins. Nat Rev Neurosci. 2012;13:605-618 doi:10.1038/nrn3302
72. Tamagnone L. Emerging role of semaphorins as major regulatory signals and potential therapeutic targets in cancer. Cancer Cell. 2012;22:145-152 doi:10.1016/j.ccr.2012.06.031
73. Takamatsu H, Kumanogoh A. Diverse roles for semaphorin-plexin signaling in the immune system. Trends Immunol. 2012;33:127-135 doi:10.1016/j.it.2012.01.008
Author contact
![]() Corresponding author: Paul H. Davis, University of Nebraska at Omaha. pdavisedu; Fax: 402-554-3532; Phone: 402-554-3379; Address: University of Nebraska at Omaha, Allwine Hall 427, 6001 Dodge Street, Omaha, NE 68182-0040
Corresponding author: Paul H. Davis, University of Nebraska at Omaha. pdavisedu; Fax: 402-554-3532; Phone: 402-554-3379; Address: University of Nebraska at Omaha, Allwine Hall 427, 6001 Dodge Street, Omaha, NE 68182-0040
Received 2017-12-6
Accepted 2018-3-17
Published 2018-4-10