ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2018; 6:98-102. doi:10.7150/jgen.26122 This volume Cite
Short Research Paper
Mitochondrial genomes of the South American frogs Eupsophus vertebralis and E. emiliopugini (Neobatrachia: Alsodidae) and their phylogenetic relationships
Elkin Y. Suárez-Villota ![]() , Camila A. Quercia, José J. Nuñez
, Camila A. Quercia, José J. Nuñez
Instituto de Ciencias Marinas y Limnológicas, Universidad Austral de Chile, Casilla 567, Valdivia, Chile.
Received 2018-3-17; Accepted 2018-5-25; Published 2018-6-19
Abstract

We report the sequencing and compare the mitochondrial genomes of the South American ground frogs Eupsophus vertebralis and E. emiliopugini and reconstruct phylogenetic relationships among Eupsophus species. These genomes consist of 16,156 and 16,711 bp in length, respectively and contain 13 protein-coding genes, two ribosomal RNA genes, 22 transfer RNA genes (tRNA), and partial non-coding D-loop region. Both genomes share 94.5% identity with 879 variable sites. A phylogenetic analysis with other available mitogenomes recovered both species as the sister clade of Alsodes gargola. Sequences from D-loop, CO1, and Cyt b, amplified and sequenced with primers developed from the mitochondrial genomes, allowed us to reconstruct phylogenetic relationships among Eupsophus species. Since our report represents the first mitogenomes for the genus Eupsophus, we expect these data will be valuable for further studies on conservation genetics and on the evolution of Patagonian amphibians.
Keywords: anurans, mitochondrial genome, Eupsophus, conservation
Introduction
The alsodid ground frogs genus Eupsophus is divided into the roseus (2n=30) and vertebralis (2n=28) groups, distributed throughout the temperate Nothofagus forests of South America [1, 2]. The roseus group is composed of E. altor, E. roseus, E. calcaratus, E. contulmoensis, E. insularis, E. septentrionalis, E. migueli, and E. nahuelbutensis, while the vertebralis group consists of E. vertebralis and E. emiliopugini [2]. The last two species are distributed mostly in Southern Chile, with marginal distribution in Argentina (Puelo Lake and Puerto Blest) [3]. Eupsophus vertebralis is included in Red List Category as Near Threatened [4], whereas E. emiliopugini as Least Concern [5]. The accelerated destruction and degradation of native Nothofagus forests, generally through fires and replacement with monocultures of exotic plant species, could render these species as vulnerable in the near future [6]. So far, no mitochondrial genomes for the genus Eupsophus have been described; here we provide mitogenomic data sets of the two species belonging to vertebralis group and reconstruct phylogenetic relationships using selected mitochondrial DNA segments from all Eupsophus species.
Adult specimens of E. vertebralis and E. emiliopugini were collected from Oncol (-39.69845, -73.32725) and Puyehue (-40,74448, -72,27309), respectively. These specimens were euthanized in accordance with Exempt Resolution No. 9244/2015 of Servicio Agrícola y Ganadero, Ministerio de Agricultura, Gobierno de Chile and included in herpetological collection from Instituto de Ciencias Marinas y Limnológicas, Univerisidad Austral de Chile (vouchers ICMLH495 and ICMLH499 for E. vertebralis and E. emiliopugini, respectively). Mitochondria were isolated from liver using the Thermo Scientific Mitochondria Isolation Kit (Cat. No. 89801) and their DNA extracted with the DNeasy Blood and Tissue Kit (Qiagen, Cat. No. 69504). The extracted DNA was used to construct an Ilumina paired-end (PE) genomic library and this library was sequenced using an Illumina MiSeq platform (Illumina, San Diego, CA). Quality control of PE reads was carried out using Trim Galore! v0.4.5 [7] and BBMap v37.90 [8]. A subset of 1.7M trimmed reads were used for reconstructing the mitochondrial genomes with MITObim v1.7 [9]. We used 12S rRNA, tRNA-Val and 16S rRNA sequences from Eupsophus altor (JX204194) and the mitochondrial genome of Telmatobius bolivianus (JF703234) as reference sequences. A total of 38,652 (for E. vertebralis) and 9,192 (for E. emiliopugini) individual mitochondrial reads were obtained with a mean coverage of 468- and 102-fold, respectively. Mitochondrial genomes were annotated in Geneious vR9 (Biomatters Ltd., Auckland, New Zealand) by aligning them to the mitogenome of T. bolivianus (JF703234) and Alsodes gargola (JX564852).
Mitogenome sizes of 16,156 and 16,711 bp for E. vertebralis and E. emiliopugini were obtained, respectively (GenBank Accession Numbers: MH070027 and MH070028). Both sequences comprise 37 genes, including 13 protein coding genes (PCGs), 22 transfer RNA (tRNA) genes, 2 ribosomal RNA (rRNA) genes, and a partial sequence from the Control Region (D-loop) (Fig. 1, Table 1). Mitogenomic base composition was A (27.9%), T (32.4%), C (19.1%), G (20.6%), and GC (39.7%) for E. vertebralis and A (29.4%), T (32.4%), C (19.1%), G (19.1%), and GC (38.2%) for E. emiliopugini. Both mitogenomes exhibited the usual mitochondrial gene order for neobatrachian frogs [10]. This order is characterized by having the Control Region located between Cyt b and a block of tRNAs (tRNAPro, tRNAGln, tRNAAla, tRNAAsn), as well as other block of tRNAs (tRNAGlu, ND6, ND5, tRNASer, and tRNAHis) located downstream from Cyt b (Fig. 1).
Mitochondrial genome structure of Eupsophus vertebralis. The same structure, consisting of 16,711 bp, was obtained for E. emiliopugini. Circular genome is shown for didactic purposes, although a segment of the Control Region remains unknown. Genes are indicated as arrows clockwise (heavy strand encoded) and arrows counter-clockwise (light strand encoded). The protein coding regions are labeled in green, tRNA genes are labeled in pink, and rRNA genes are labeled in red colors. GC (blue) and AT (green) content are shown.
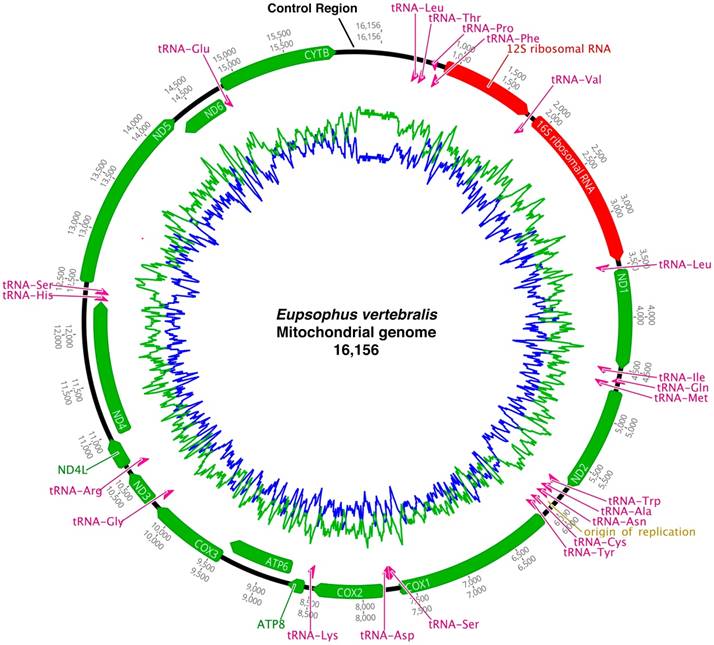
Organization of the mitochondrial genomes of Eupsophus vertebralis and E. emiliopugini. Strand, start codon, and gaps+/overlaps are identical for both species. Variable sites corresponding to the number of bases pairs do not coincide between both species.
| Gene | E. vertebralis | E. emiliopugini | Strand | Start codon | Stop codon | Gaps+/overlaps (nt) | Variable sites | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| From | To | Size | From | To | Size | ||||||
| Control | 1 | 598 | 1 | 1,171 | L | 0 | 712 | ||||
| region | 16004 | 16156 | 16,575 | 16,711 | |||||||
| tRNALeu1 | 599 | 670 | 72 | 1,172 | 1,243 | 72 | H | 0 | 1 | ||
| tRNAThr | 671 | 743 | 73 | 1,244 | 1,316 | 73 | H | 0 | 0 | ||
| tRNAPro | 743 | 811 | 69 | 1,316 | 1,384 | 69 | L | 0 | 0 | ||
| tRNAPhe | 811 | 879 | 69 | 1,384 | 1,451 | 68 | H | 0 | 3 | ||
| 12S | 880 | 1,809 | 930 | 1,452 | 2,380 | 929 | H | 0 | 3 | ||
| tRNAVal | 1,810 | 1,878 | 69 | 2,381 | 2,449 | 69 | H | 0 | 0 | ||
| 16S | 1,879 | 3,473 | 1,595 | 2,450 | 4,044 | 1,595 | H | 0 | 10 | ||
| tRNALeu2 | 3,474 | 3,546 | 73 | 4,045 | 4,117 | 73 | H | 0 | 0 | ||
| ND1 | 3,547 | 4,507 | 961 | 4,118 | 5,078 | 961 | H | ? | T* | 0 | 12 |
| tRNAIle | 4,508 | 4,578 | 71 | 5,079 | 5,149 | 71 | H | 0 | 3 | ||
| tRNAGln | 4,578 | 4,648 | 71 | 5,149 | 5,219 | 71 | L | -1 | 0 | ||
| tRNAMet | 4,648 | 4,716 | 69 | 5,219 | 5,287 | 69 | H | 0 | 0 | ||
| ND2 | 4,717 | 5,750 | 1,034 | 5,288 | 6,321 | 1,034 | H | ATT | TA* | 0 | 14 |
| tRNATrp | 5,751 | 5,820 | 70 | 6,322 | 6,391 | 70 | H | 0 | 1 | ||
| tRNAAla | 5,821 | 5,889 | 69 | 6,392 | 6,460 | 69 | L | 0 | 1 | ||
| tRNAAsn | 5,890 | 5,962 | 73 | 6,461 | 6,533 | 73 | L | 27 | 0 | ||
| tRNACys | 5,989 | 6,051 | 63 | 6,560 | 6,622 | 63 | L | 0 | 0 | ||
| tRNATyr | 6,052 | 6,121 | 70 | 6,623 | 6,692 | 70 | L | 2 | 0 | ||
| CO1 | 6,123 | 7,667 | 1,545 | 6,694 | 8,238 | 1,545 | H | GTG | AGG | 0 | 13 |
| tRNASer2 | 7,668 | 7,737 | 70 | 8,239 | 8,308 | 70 | L | 2 | 0 | ||
| tRNAAsp | 7,739 | 7,807 | 69 | 8,310 | 8,378 | 69 | H | 2 | 0 | ||
| CO2 | 7,809 | 8,496 | 688 | 8,380 | 9,067 | 688 | H | ATG | T* | 0 | 11 |
| tRNALys | 8,497 | 8,568 | 72 | 9,068 | 9,139 | 72 | H | 0 | 0 | ||
| ATP8 | 8,569 | 8,733 | 165 | 9,140 | 9,304 | 165 | H | ATG | TAA | -9 | 1 |
| ATP6 | 8,724 | 9,407 | 684 | 9,295 | 9,978 | 684 | H | ATG | TAA | 0 | 10 |
| CO3 | 9,407 | 10,190 | 784 | 9,978 | 10,761 | 784 | H | ATG | T* | 0 | 13 |
| tRNAGly | 10,191 | 10,259 | 69 | 10,762 | 10,830 | 69 | H | 0 | 0 | ||
| ND3 | 10,260 | 10,599 | 340 | 10,831 | 11,170 | 340 | H | ATG | T* | 0 | 2 |
| tRNAArg | 10,600 | 10,668 | 69 | 11,171 | 11,239 | 69 | H | 6 | 0 | ||
| ND4L | 10,674 | 10,973 | 300 | 11,245 | 11,544 | 300 | H | ATG | TAA | -6 | 5 |
| ND4 | 10,967 | 12,331 | 1,365 | 11,538 | 12,902 | 1,365 | H | ATG | TAG | 0 | 21 |
| tRNAHis | 12,332 | 12,399 | 68 | 12,903 | 12,970 | 68 | H | 0 | 1 | ||
| tRNASer1 | 12,400 | 12,466 | 67 | 12,971 | 13,037 | 67 | H | 38 | 0 | ||
| ND5 | 12,504 | 14,306 | 1,803 | 13,075 | 14,877 | 1,803 | H | ATG | AGA | -16 | 28 |
| ND6 | 14,290 | 14,784 | 495 | 14,861 | 15,355 | 495 | L | ATG | AGA | 0 | 0 |
| tRNAGlu | 14,785 | 14,852 | 68 | 15,356 | 15,423 | 68 | L | 3 | 1 | ||
| Cyt b | 14,855 | 16,003 | 1,149 | 15,426 | 16,574 | 1,149 | H | ATG | TAG | 13 | |
? start codon not determined
* TAA stop codon is completed by the addition of 3' A residues to the mRNA
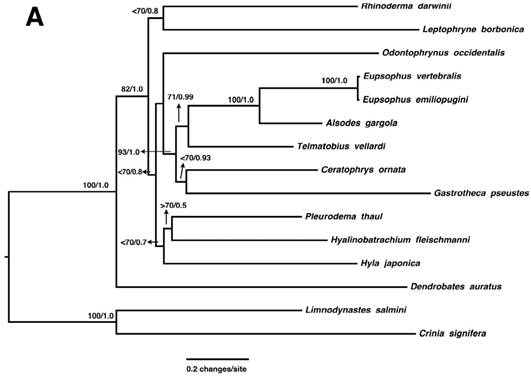
The phylogenetic position of E. vertebralis and E. emiliopugini was evaluated through Maximum Likelihood (ML) and Bayesian approaches using previously published mitogenome sequences of neobatrachians related to Alsodidae [11]. Mitogenomes of Limnodynastes salmini and Crinia signifera (Myobatrachidae) were used as outgroup [11]. Sequences of the two rRNAs, the concatenated tRNAs, and 13 protein-coding genes were aligned using Muscle [12] and Clustal W [13], implemented in Geneious. The best-fitting partitioning schemes and models of nucleotide substitution were selected using the Bayesian information criterion, implemented in PartitionFinder v1.1.0 [14]. ML analysis was performed using GARLI v2.0 with 1000 bootstrap replicates [15]. Both ML and Bayesian inference, performed in MrBayes v3.04b [16] and run for 1.0 x 107 generations sampled every 1000 steps, showed E. vertebralis and E. emiliopugini as a clade (Fig. 2A). This clade was related to Alsodes gargola, from the same family (Alsodidae) [2], as expected (Fig. 2A).
A. Phylogenetic relationships of neobatrachian species inferred from mitochondrial genomes, including the mitogenomes of Eupsophus vertebralis and E. emiliopugini. B. Phylogenetic relationships of Eupsophus species inferred from concatenated D-loop, CO1, and Cyt b markers. Both A and B, Maximum likelihood (ML) and Bayesian analyses depicted the same topology. ML bootstrap percentage /Bayesian posterior probabilities are shown at nodes.
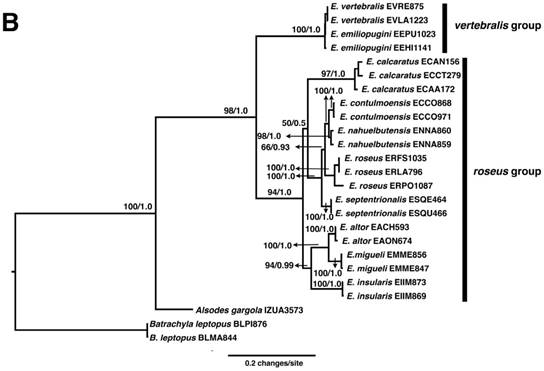
We used a combination of primers previously reported [17-19] and primers whose design was based on the mitochondrial genomes here reported (Cyt b F1 5'-AGCTACTGCAATCAACCCCC-3', Cyt b F2 5'-AATGCACTACACTGCCGACA-3' Cyt b R1 5'-GCTAAGACGCCTCCCAGTTT-3'), to amplify and sequence segments of D-loop, CO1, and Cyt b from the ten Eupsophus species (GenBank Accession Numbers: MH301116-MH301181). Phylogenetic analysis was carried out using Garli and MrBayes as described above. Sequences from Batrachyla leptopus and A. gargola were used as outgroup. The phylogenetic relationships among Eupsophus species were congruent with previous hypotheses [1, 3], showing two clades corresponding to vertebralis and roseus groups (Fig. 2B).
This is the first report of mitogenomes of species in the genus Eupsophus. In this genus, seven of 10 species are included in the IUCN Red List, namely, E. insularis (Critically Endangered), E. queulensis (Vulnerable), E. migueli, E. contulmoensis, and E. nahuelbutensis (Endangered), and E. vertebralis and E. roseus (Near Threatened). Our data provides a baseline for further studies on these endemic, threatened and poorly known species.
Acknowledgements
We thank to Andrea Silva of the Austral-omics, Universidad Austral de Chile, for technical assistance. We also thank Milton H. Gallardo for proofreading the manuscript. The Corporacion Nacional Forestal, Ministerio de Agricultura, Gobierno de Chile allows to collect buccal swabs samples of Eupsophus species from wild protected areas (Permit No. 11/2016.-CPP/MDM/jcr/29.02.2016). Fondecyt 3160328 to EYS-V and DID-UACh 2014-16 to JJN supported this research.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Formas JR, Brieva L. Immunological relationships of the South American frog genus Eupsophus (Leptodactylidae). Biochem Syst Ecol. 1992;20:747-51
2. Frost DR. Amphibian species of the world: an online reference. Version 6.0. New York, American Museum of Natural History http://research.amnh.org/herpetology/amphibia/index.html. Accessed 10 May. 2018
3. Blotto BL, Nunez JJ, Basso NG, Ubeda CA, Wheeler WC, Faivovich J. Phylogenetic relationships of a Patagonian frog radiation, the Alsodes + Eupsophus clade (Anura: Alsodidae), with comments on the supposed paraphyly of Eupsophus. Cladistics. 2013;29:113-31
4. Veloso A, Núñez H, Núñez JJ, Ortiz JC, Úbeda C. Eupsophus vertebralis. The IUCN Red List of Threatened Species. http://dx.doi.org/10.2305/IUCN.UK.2010-2.RLTS.T57080A11566149.en. Accessed 10 May. 2018
5. Úbeda C, Veloso A, Nuñez H, Formas JR, Basso NG. Eupsophus emiliopugini. The IUCN Red List of Threatened Species. http://dx.doi.org/10.2305/IUCN.UK.2010-2.RLTS.T57077A11565131.en. Accessed 10 May. 2018
6. Stuart SN, Hoffmann M, Chanson JS, Cox NA, Berridge RJ, Ramani P. et al. Threatened Amphibians of the World. Barcelona: Lynx Edicions. 2008
7. Krueger F. Trim Galore!: a wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/. Accessed 10 May. 2018
8. Bushnell B. BBMap. https://sourceforge.net/projects/bbmap/. Accessed 10 May. 2018
9. Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 2013;41:e129
10. Irisarri I, Mauro DS, Abascal F, Ohler A, Vences M, Zardoya R. The origin of modern frogs (Neobatrachia) was accompanied by acceleration in mitochondrial and nuclear substitution rates. BMC Genomics. 2012;13:626
11. Zhang P, Liang D, Mao RL, Hillis DM, Wake DB, Cannatella DC. Efficient sequencing of anuran mtDNAs and a mitogenomic exploration of the phylogeny and evolution of frogs. Mol Biol Evol. 2013;30:1899-915
12. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792-7
13. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673-80
14. Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29:1695-701
15. Bazinet AL, Zwickl DJ, Cummings MP. A gateway for phylogenetic analysis powered by grid computing featuring GARLI 2.0. Syst Biol. 2014;63:812-8
16. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572-4
17. Degnan SM, Moritz C. Phylogeography of mitochondrial DNA in two species of white-eyes in Australia. The Auk. 1992;109:800-11
18. Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294-9
19. Goebel AM, Donnelly JM, Atz ME. PCR primers and amplification methods for 12S ribosomal DNA, the control region, cytochrome oxidase I, and cytochrome b in bufonids and other frogs, and an overview of PCR primers which have amplified DNA in amphibians successfully. Mol Phylogenet Evol. 1999;11:163-99
Author contact
![]() Corresponding author: esuarezvcom
Corresponding author: esuarezvcom