ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2019; 7:18-25. doi:10.7150/jgen.32194 This volume Cite
Research Paper
Complete Genome Sequence of Arthrobacter sp. Strain MN05-02, a UV-Resistant Bacterium from a Manganese Deposit in the Sonoran Desert
Konosuke Mark Ii1,2, Nobuaki Kono1,3, Ivan Glaucio Paulino-Lima4, Masaru Tomita1,2,3, Lynn Justine Rothschild5, Kazuharu Arakawa1,2,3 ![]()
1. Institute for Advanced Biosciences, Keio University, Tsuruoka, Yamagata, 997-0052, Japan
2. Faculty of Environment and Information Studies, Keio University, Yamagata, 997-0052, Japan
3. Graduate School of Media and Governance, Keio University, Yamagata, 997-0052, Japan
4. Blue Marble Space Institute of Science at NASA Ames Research Center, Mountain View, CA, USA, 94035-0001
5. NASA Ames Research Center, Moffett Field, CA, USA, 94035-0001
Received 2018-12-18; Accepted 2019-1-8; Published 2019-2-8
Abstract

Arthrobacter sp. strain MN05-02 is a UV-resistant bacterium isolated from a manganese deposit in the Sonoran Desert, Arizona, USA. The LD10 of this strain is 123 Jm-2, which is twice that of Escherichia coli, and therefore can be a useful resource for comparative study of UV resistance and the role of manganese on this phenotype. Its complete genome is comprised of a chromosome of 3,488,433 bp and a plasmid of 154,991 bp. The chromosome contains 3,430 putative genes, including 3,366 protein coding genes, 52 tRNA and 12 rRNA genes. Carotenoid biosynthesis operon structure coded within the genome mirrors the characteristic orange-red pigment this bacterium produces, which presumably partly contribute to its UV resistance.
Keywords: Actinobacteria, UV-resistance, orange-red pigment, carotenoid biosynthesis, Sonoran Desert
Introduction
Many microorganisms within the category of extremophiles are known to survive intense ultraviolet (UV) radiation or high dose of ionizing radiation, while such natural environment is lacking on Earth. Through the studies of these bacteria including the radio-tolerant model Deinococcus radiodurans, radio- tolerance is speculated to be the by-product of desiccation tolerance [1]. Defense and repair mechanisms of such tolerance is likely multi-parametric and are diverse among different organisms, but the central damage is the oxidative injury of DNA and proteins [2]. Recently, removal mechanism of the reactive oxygen species (ROS) by Mn2+ complex has been proven by multiple models including Deinococcus radiodurans, and is proposed to be a key strategy in ROS defense in microorganisms [3]. Correlation of radio-resistance and intracellular Mn/Fe ratio was also reported among various bacterial species [4].
In order to survey a wide range of radiation- resistant bacteria and to elucidate the contribution of Mn to their tolerance, we have previously reported a comprehensive screening of UV-tolerant bacteria from a manganese deposit in the Sonoran Desert, Arizona, which is considered a Mars analog due to its extreme dryness and intense solar UV radiation [5]. In this report, we describe the complete genome sequencing of one of the isolates in the Arthrobacter genus of Micrococcaceae family designated strain MN05-02. Arthrobacter and its closely related genus Kocuria were the two most dominant groups found in the above screening with quite diverse lethal dose of UV-C. Therefore, a comparative genomic study of these species would possibly unveil the genetic mechanisms contributing to radio-resistance in Micrococcaceae. A unique trait of these groups is the orange-red pigmentation of colonies, which presumeably contributes to the defense against UV. We highlight the biosynthesis pathways producing these characteristic pigments from our genomic study.
Materials and Methods
Sequenced strain
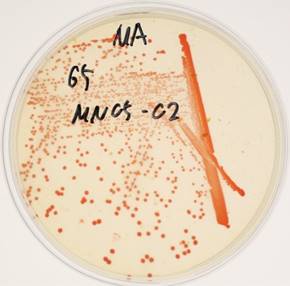
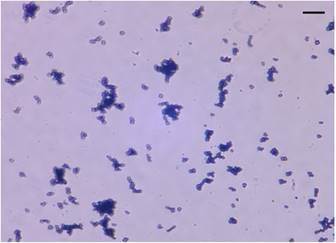
Arthrobacter sp. strain MN05-02 was isolated from a manganese deposit in the Sonoran Desert, Arizona, USA. In order to screen UV-resistant bacteria, sampled soil was sprinkled over Marine Agar 2216 (Difco) plates under sterile conditions, and was exposed to UV-C in a UV radiation hood containing two germicidal lamps for 4 kJm-2, and surviving colonies were picked. The LD10 of this strain is 123 kJm-2, which is twice that of Escherichia coli. Arthrobacter sp. strain MN05-02 produces characteristic orange-red colored colony (Fig. 1), which presumably contributes to its UV-tolerance. Arthrobacter sp. strain MN05-02 grows best in Marine Broth 2216 (Difco), suggesting its preference for high salt content in the medium. Optimal temperature is around 30 ˚C, and this strain is aerobic, non-pathogenic, and free-living. Cells are spherical in shape, and about 1µm in diameter (Fig. 2).
Colonies of Arthrobacter sp. strain MN05-02 on agarose plate with characteristic orange-red pigment.
Stereo micrograph of Arthrobacter sp. strain MN05-02 (x2000 magnification). Scale bar (upper-right) is 10µm.
Growth conditions and genomic DNA preparation
Arthrobacter sp. Strain MN05-02 was cultivated in Marine Broth 2216 (Difco) at 30 ˚C. For Illumina sequencing, cell pellets were homogenized with zirconia beads in Multi Beads Shocker (Yasui Kikai), and genomic DNA was extracted using DNeasy Kit (Qiagen). After purification with AMPure XP beads (Beckman-Coulter), DNA was fragmented to 800 bp using Covaris M220, and Illumina library was prepared with HyperPlus Kit (KAPA) without enzymatic fragmentation.
For the nanopore sequencing library, genomic DNA was extracted and purified using a conventional liquid isolation method (Saito and Miura, 1963, PMID: 14071565) with some modifications. Briefly, cells were harvested by centrifugation at 4,000 rpm for 10 min at 4 °C with a 14 ml round bottom tube. The pellet was suspended in 700 µl of Sucrose-Lysozyme buffer (560 µl of 25 % sucrose-TES, 80 µl of 0.2 M EDTA, and 640 µg of Lysozyme). After incubation at 37 °C for 30 min, 70 µl of Proteinase K (20 mg ml-1) was added with additional incubation at 37°C for 30 min. The resultant was homogenized with 770 µl of SDS-TE buffer (70 µl of 10% SDS and 700 µl of TE) by gentle inversion. The homogenate was extracted with 1 ml of phenol by extremely gentle inversion, centrifuged at 4,000 rpm for 10 min, and the aqueous phase transferred into a fresh 14 ml round bottom tube. To precipitate genomic DNA, 3 ml of ethanol was added, and the precipitate was transferred into a new 1.5 ml tube. After a 70% ethanol wash, the precipitate was dried at room temperature for a few minutes and re-suspended in 400 µl of TE buffer containing 10 µg of RNase A, then incubated at room temperature overnight with gentle agitation. Ten micrograms of purified DNA was size selected using Blue Pippin (Sage Science) 0.75% Gel Cassette with Marker S1 High-Pass mode over 10 kbp, and fragment size was confirmed using TapeStation Genomic ScreenTape (Agilent). One microgram of size-selected genomic DNA was used to prepare a sequencing library using Ligation Sequencing Kit 1D (SQK-LSK108, Oxford Nanopore Technologies), omitting the optional fragmentation and repair steps. The resulting library quality was checked using TapeStation Genomic ScreenTape (Agilent).
Genome sequencing, assembly, and annotations
Illumina library was sequenced on MiSeq (Illumina) with v3 600 cycles kit in multiplexed paired-ends, yielding 220 Mbp roughly corresponding to x60 coverage. The nanopore library was sequenced on a MinION device with R9.4 flowcell (Oxford Nanopore) for 24 hours with live basecalling on MinKNOW v.1.5.18, yielding 59,428 reads totaling 374 Mbp (around x100 coverage), where read N50 was 18,475 bp. Resulting nanopore reads were assembled with Canu 1.6 with default parameters [6], resulting in two contigs with length 3,449,825 bp and 175,137 bp, respectively. Both contigs were suggested be circular by the Canu software, but we further confirmed the circularity of the contigs by aligning the raw nanopore reads. The shorter contig was identified to be a plasmid by BLAST search on NCBI. The resulting complete chromosome and plasmid sequences were polished using Pilon with Illumina reads, resulting in the final 3,488,433bp and 154,991bp genomes.
The genome and plasmid sequences were annotated using the DFAST pipeline[7]. DFAST internally uses MGA tool for coding sequence prediction[8], barmap for rRNA genes (https://github.com/tseemann/barrnap), Aragorn for tRNA genes [9]. The predicted coding regions are then queried against DFAST database.
Results and Discussion
The genome was initially sequenced with the Illumina MiSeq instrument at Keio University, Tsuruoka City, Japan, yielding 368,916 paired-end reads roughly corresponding x60 coverage. The genome was further sequenced with an Oxford Nanopore Technologies MinION device to complete the gaps. Sequencing was performed in the conference venue of the 5th NGS-Field Meeting in Sendai, Japan (http://ngs5.org), yielding a total of 375 Mbp (around x100 coverage) with read N50 length of 18kbp. The project information including accession numbers is summarized in Table 2.
Classification and general features of Arthrobacter sp. strain MN05-02 according to the MIGS recommendation [16]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | IDA, TAS [5] | |
| Phylum Actinobacteria | IDA, TAS [5] | ||
| Class Actinobacteria | IDA, TAS [5] | ||
| Order Actinomycetales | IDA, TAS [5] | ||
| Family Micrococcaceae | IDA, TAS [5] | ||
| Genus Arthrobacter | IDA, TAS [5] | ||
| Species sp. | IDA, TAS [5] | ||
| strain: MN05-02 | |||
| Gram stain | Positive | IDA | |
| Cell shape | coccus | IDA | |
| Motility | N/A | ||
| Sporulation | N/A | ||
| Temperature range | N/A | ||
| Optimum temperature | 30°C | TAS [5] | |
| pH range; Optimum | N/A | ||
| Carbon source | Monosaccharides | TAS [5] | |
| MIGS-6 | Habitat | Desert surface soil | TAS [5] |
| MIGS-6.3 | Salinity | N/A | |
| MIGS-22 | Oxygen requirement | Aerobic | TAS [5] |
| MIGS-15 | Biotic relationship | free-living | NAS |
| MIGS-14 | Pathogenicity | non-pathogen | NAS |
| MIGS-4 | Geographic location | Sonoran Desert, Arizona, USA | TAS [5] |
| MIGS-5 | Sample collection | 2011 | TAS [5] |
| MIGS-4.1 | Latitude | 34°20′13.734″N | TAS [5] |
| MIGS-4.2 | Longitude | 113°37′33.666″W | TAS [5] |
| MIGS-4.4 | Altitude | 550 m | TAS [5] |
a Evidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [17]
Project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Complete |
| MIGS-28 | Libraries used | Nanopore 1D Ligation library and Illumina paired-end libraries in size of 800 bp |
| MIGS 29 | Sequencing platforms | MinION (Oxford Nanopore Technologies) and MiSeq (Illumina) |
| MIGS 31.2 | Fold coverage | x100 (MinION), x60 (MiSeq) |
| MIGS 30 | Assemblers | Canu 1.6 |
| MIGS 32 | Gene calling method | MGA in DFAST |
| Locus Tag | MN0502 | |
| Genbank ID | AP018697-AP018698 | |
| GenBank Date of Release | ||
| GOLD ID | N/A | |
| BIOPROJECT | PRJDB7048 | |
| MIGS 13 | Source Material Identifier | |
| Project relevance | Biotechnology, evolution |
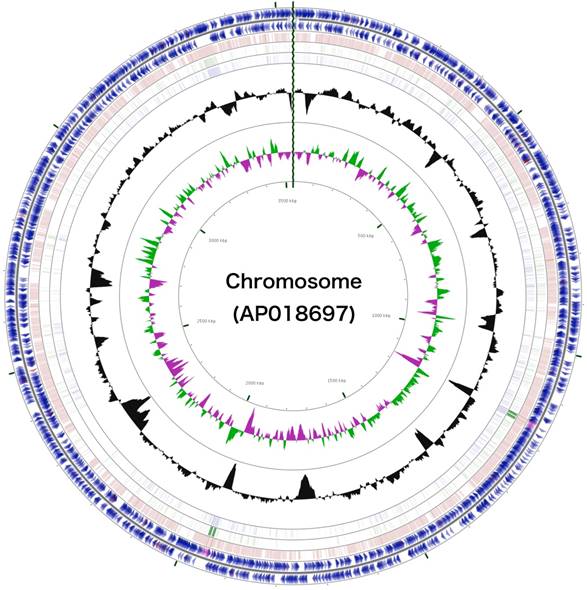
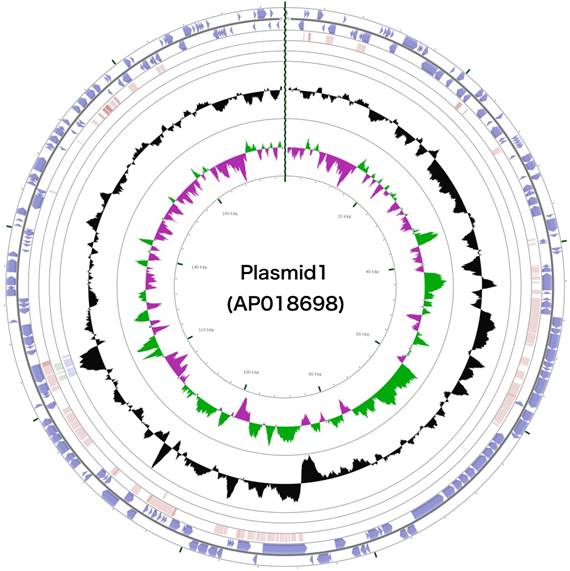
The 3,488,433 bp chromosome (Fig. 4) and 154,991 bp plasmid (Fig. 5) of Arthrobacter sp. strain MN05-02 had GC content of 69.11% and 62.16%, respectively. They contained 3,430 and 177 putative genes, with 3,366 coding sequences, 52 rRNA genes, 12 tRNA genes in the chromosome, and 177 coding sequences and no RNA genes in the plasmid. Seventy four percent of the predicted genes were assigned to one of 25 COG categories, but 27.28% remained unannotated. The distribution of COG functional categories is shown in Table 4.
Schematic representation of the complete chromosome sequence of Arthrobacter sp. strain MN05-02. From the outmost circle, tracks represent (1) CDS (blue), tRNA (orange) and rRNA (violet) on forward strand, (2) CDS (blue), tRNA (orange) and rRNA (violet) on reverse strand, (3) BLAST hits (e<1e-15) to A. agilis, (4) BLAST hits (e<1e-15) to A. alpinus, (5) BLAST hits (e<1e-15) to A. castelli, (6) GC content, (7) GC skew (green: positive, purple: negative). GCView [21] was used to create this genome map.
Schematic representation of the complete plasmid sequence of Arthrobacter sp. strain MN05-02. From the outmost circle, tracks represent (1) CDS (blue), tRNA (orange) and rRNA (violet) on forward strand, (2) CDS (blue), tRNA (orange) and rRNA (violet) on reverse strand, (3) BLAST hits (e<1e-15) to A. agilis, (4) BLAST hits (e<1e-15) to A. alpinus, (5) BLAST hits (e<1e-15) to A. castelli, (6) GC content, (7) GC skew (green: positive, purple: negative). GCView [21] was used to create this genome map.
Summary of genome: one chromosome and one plasmid
| Label | Size (Mb) | Topology | INSDC identifier | RefSeq ID |
|---|---|---|---|---|
| Chromosome | 3.488 | Circular | AP018697 | |
| Plasmid 1 | 0.155 | Circular | AP018698 |
Genome statistics.
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 3,488,433 | 100.00 |
| DNA coding (bp) | 3,061,318 | 87.76 |
| DNA G+C (bp) | 2,410,932 | 69.11 |
| DNA scaffolds | 1 | - |
| Total genes | 3,430 | 100.00 |
| Protein coding genes | 3,366 | 98.13 |
| RNA genes | 64 | 1.86 |
| Pseudo genes | - | - |
| Genes in internal clusters | 769 | 22.41 |
| Genes with function prediction | 2638 | 76.90 |
| Genes assigned to COGs | 2491 | 72.62 |
| Genes with Pfam domains | 2420 | 70.55 |
| Genes with signal peptides | 376 | 10.96 |
| Genes with transmembrane helices | 818 | 23.84 |
| CRISPR repeats | 0 | 0 |
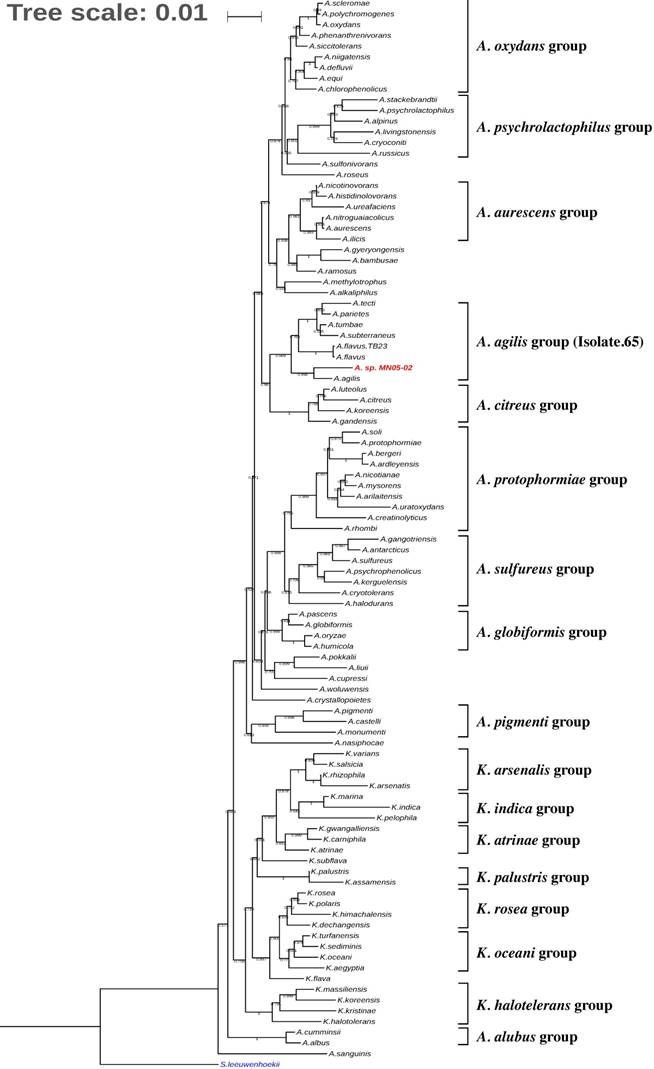
Fig. 3 shows the phylogenetic placement of Arthrobacter sp. Strain MN05-02 based on 16S rRNA maximum likelihood phylogenetic tree. The closest sequenced relative is A. agilis, which is reported to produce pink pigment comprised of dimethylhexadecylamine and carotenoids. Detailed analysis of the gene content and the synteny of carotenoid biosynthesis pathways, however, show divergence between the two species.
Phylogenetic tree indicating the position of Arthrobacter sp. strain MN05-02 (in red) relative to other genomes sequenced within the genus Arthrobacter and Kocuria. Streptomyces leeuwenhoekii was used as the outgroup. The tree was inferred from 16S rRNA sequences using MAFFT v.7.312 as aligner [18] and with FastTree 2.1 (Maximum-likelihood) [19]. Tree was visualized with iTOL v4[20]. Numbers at the branches indicate the bootstrap values (N=100).
Many bacteria of the family Micrococcus are known to produce carotenoid pigments, and details are known the carotenoid biosynthesis gene cluster from Micrococcus luteus [10]. Based on these set of genes and on the orthologs in Corynebacterium glutamicum [11] and Dietzia sp [12], we identified the carotenoid biosynthesis gene cluster in the MN05-02 strain (Fig. 6).
Schematic representation of the carotenoid biosynthesis pathways of Corynebacterium glutamicum, Dietzia sp CQ4, Micrococcus luteus, Arthrobacter castelli, A alpinus, A. agilis, and A. sp. strain MN05-02. Gene upstream of the carotenoid biosynthesis cascade crtE, crtB, crtI, and crtEb are conserved throughout, suggesting the production of C40/C45 carotenoids, but further production and modification of C50 carotenoids (crtY and crtX) seem to be lacking in A. agilis and A. sp. MN05-02.
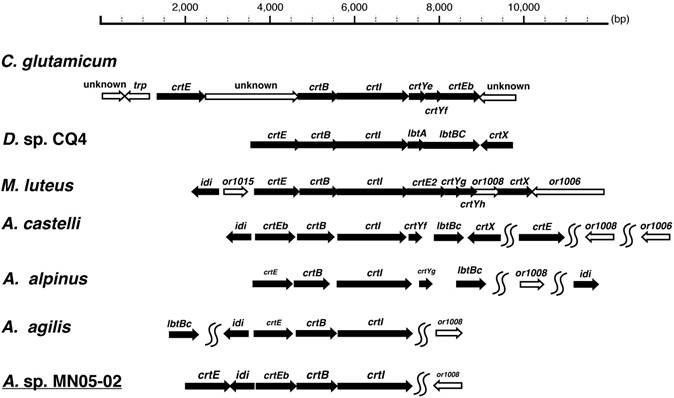
Comparison of the synteny of carotenoid biosynthesis genes of Arthrobacter sp. strain MN05-02 as well as three related species (Arthrobacter agilis [13], Arthrobacter alpinus [14], and Arthrobacter castelli [15]) is shown in Fig. 6. A clear difference of A. agilis, and Arthrobacter sp. strain MN05-02 from others is the lack of C45/C50 cyclase crtY and C.p.450 glucosyltransferase crtX. These species are therefore likely to produce C40 lycopene-derivatives unlike other species producing C45/C50 carotenoids. These gene conservation patterns mirror the colony color, where A. agilis, and Arthrobacter sp. strain MN05-02 have orange-red appearance whereas other species exhibit yellow pigmentation.
In this study, we sequenced and assembled the complete genome and plasmid of the UV-resistant bacterium Arthrobacter sp. strain MN05-02, isolated from the surface soil of Sonoran Desert, Arizona, USA. Characteristic orange-red pigment produced by this species presumably contribute, at least in part, to its UV-resistance, and the genomic basis for the production of this pigment is confirmed by comparing the carotenoid biosynthesis operon structures with other bacteria.
Number of genes associated with general COG functional categories.
| Code | Value | %age | Description |
|---|---|---|---|
| J | 150 | 4.23 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.02 | RNA processing and modification |
| K | 189 | 5.33 | Transcription |
| L | 129 | 3.64 | Replication, recombination and repair |
| B | 1 | 0.02 | Chromatin structure and dynamics |
| D | 22 | 0.62 | Cell cycle control, Cell division, chromosome partitioning |
| V | 29 | 0.81 | Defense mechanisms |
| T | 114 | 3.21 | Signal transduction mechanisms |
| M | 128 | 3.61 | Cell wall/membrane biogenesis |
| N | 30 | 0.84 | Cell motility |
| U | 32 | 0.90 | Intracellular trafficking and secretion |
| O | 82 | 2.31 | Posttranslational modification, protein turnover, chaperones |
| C | 141 | 3.97 | Energy production and conversion |
| G | 280 | 7.90 | Carbohydrate transport and metabolism |
| E | 249 | 7.02 | Amino acid transport and metabolism |
| F | 73 | 2.06 | Nucleotide transport and metabolism |
| H | 109 | 3.07 | Coenzyme transport and metabolism |
| I | 91 | 2.56 | Lipid transport and metabolism |
| P | 111 | 3.13 | Inorganic ion transport and metabolism |
| Q | 51 | 1.43 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 288 | 8.12 | General function prediction only |
| S | 191 | 5.39 | Function unknown |
| - | 1052 | 29.69 | Not in COGs |
The total is based on the total number of protein coding genes in the genome.
Abbreviations
BLAST: Basic local alignment search tool; COG: Clusters of Orthologous Groups; MGA: MetaGeneAnnotator; MIGS: Minimum information on the genome sequence; ROS: Reactive Oxygen Species; UV: Ultraviolet radiation.
Acknowledgements
The authors thank Nozomi Abe, Yuki Onozawa, and Yuki Takai for technical support. This work is supported by the Astrobiology Center Program of National Institutes of Natural Sciences (NINS) (Grant Number AB281019 and AB291020), and in part by research funds from the Yamagata Prefectural Government and Tsuruoka City, Japan. IGPL's fellowship was provided by the NASA Postdoctoral Program.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mattimore V, Battista JR. Radioresistance of Deinococcus radiodurans: functions necessary to survive ionizing radiation are also necessary to survive prolonged desiccation. J Bacteriol. 1996;178:633-7
2. Pavlopoulou A, Savva GD, Louka M, Bagos PG, Vorgias CE, Michalopoulos I, Georgakilas AG. Unraveling the mechanisms of extreme radioresistance in prokaryotes: Lessons from nature. Mutat Res Rev Mutat Res. 2016;767:92-107
3. Culotta VC, Daly MJ. Manganese complexes: diverse metabolic routes to oxidative stress resistance in prokaryotes and yeast. Antioxid Redox Signal. 2013;19:933-44
4. Daly MJ. A new perspective on radiation resistance based on Deinococcus radiodurans. Nat Rev Microbiol. 2009;7:237-45
5. Paulino-Lima IG, Fujishima K, Navarrete JU, Galante D, Rodrigues F, Azua-Bustos A, Rothschild LJ. Extremely high UV-C radiation resistant microorganisms from desert environments with different manganese concentrations. J Photochem Photobiol B. 2016;163:327-36
6. Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722-36
7. Tanizawa Y, Fujisawa T, Nakamura Y. DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics. 2018;34:1037-9
8. Noguchi H, Taniguchi T, Itoh T. MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008;15:387-96
9. Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11-6
10. Netzer R, Stafsnes MH, Andreassen T, Goksoyr A, Bruheim P, Brautaset T. Biosynthetic pathway for gamma-cyclic sarcinaxanthin in Micrococcus luteus: heterologous expression and evidence for diverse and multiple catalytic functions of C(50) carotenoid cyclases. J Bacteriol. 2010;192:5688-99
11. Krubasik P, Kobayashi M, Sandmann G. Expression and functional analysis of a gene cluster involved in the synthesis of decaprenoxanthin reveals the mechanisms for C50 carotenoid formation. Eur J Biochem. 2001;268:3702-8
12. Tao L, Yao H, Cheng Q. Genes from a Dietzia sp. for synthesis of C40 and C50 beta-cyclic carotenoids. Gene. 2007;386:90-7
13. Singh RN, Gaba S, Yadav AN, Gaur P, Gulati S, Kaushik R, Saxena AK. First high quality draft genome sequence of a plant growth promoting and cold active enzyme producing psychrotrophic Arthrobacter agilis strain L77. Stand Genomic Sci. 2016;11:54
14. See-Too WS, Ee R, Lim YL, Convey P, Pearce DA, Mohidin TBM, Yin WF, Chan KG. Complete genome of Arthrobacter alpinus strain R3.8, bioremediation potential unraveled with genomic analysis. Stand Genomic Sci. 2017;12:52
15. Heyrman J, Verbeeren J, Schumann P, Swings J, De Vos P. Six novel Arthrobacter species isolated from deteriorated mural paintings. Int J Syst Evol Microbiol. 2005;55:1457-64
16. Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, Ashburner M, Axelrod N, Baldauf S, Ballard S, Boore J, Cochrane G, Cole J, Dawyndt P, De Vos P, DePamphilis C, Edwards R, Faruque N, Feldman R, Gilbert J, Gilna P, Glockner FO, Goldstein P, Guralnick R, Haft D, Hancock D, Hermjakob H, Hertz-Fowler C, Hugenholtz P, Joint I, Kagan L, Kane M, Kennedy J, Kowalchuk G, Kottmann R, Kolker E, Kravitz S, Kyrpides N, Leebens-Mack J, Lewis SE, Li K, Lister AL, Lord P, Maltsev N, Markowitz V, Martiny J, Methe B, Mizrachi I, Moxon R, Nelson K, Parkhill J, Proctor L, White O, Sansone SA, Spiers A, Stevens R, Swift P, Taylor C, Tateno Y, Tett A, Turner S, Ussery D, Vaughan B, Ward N, Whetzel T, San Gil I, Wilson G, Wipat A. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541-7
17. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25-9
18. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017
19. Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641-50
20. Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242-5
21. Grin I, Linke D. GCView: the genomic context viewer for protein homology searches. Nucleic Acids Res. 2011;39:W353-6
Author contact
![]() Corresponding author: Kazuharu Arakawa, Institute for Advanced Biosciences, Keio University, Mizukami 246-2, Kakuganji, Tsuruoka, Yamagata, 997-0052, Japan. E-mail: gaoukeio.ac.jp
Corresponding author: Kazuharu Arakawa, Institute for Advanced Biosciences, Keio University, Mizukami 246-2, Kakuganji, Tsuruoka, Yamagata, 997-0052, Japan. E-mail: gaoukeio.ac.jp