ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2017; 5:4-11. doi:10.7150/jgen.18378 This volume Cite
Research Paper
Detection of Intestinal Pathogens in River, Shore, and Drinking Water in Lima, Peru
David C. Grothen†, Sydney J. Zach†, Paul H. Davis ![]()
Department of Biology, University of Nebraska at Omaha, Omaha NE 68182-0040, USA
† These authors contributed equally.
Published 2017-1-17
Abstract
Water quality management is an ongoing struggle for many locations worldwide. Current testing of water supplies can be time-consuming, expensive, and lack sensitivity. This study describes an alternative, easy-to-use, and inexpensive method to water sampling and testing at remote locations. This method was employed to detect a number of intestinal pathogens in various locations of Lima, Peru. A total of 34 PCR primer pairs were tested for specificity and high-yield amplification for 12 different pathogens using known DNA templates. Select primers for each pathogen were then tested for minimum detection limits of DNA. Water samples were collected from 22 locations. PCR was used to detect the presence of a pathogen, virulence factors, or differentiate between pathogenic species. In 22 water samples, cholera toxin gene was detected in 4.5% of samples, C. perfringens DNA was detected in 50% of samples, E. histolytica DNA was detected in 54.5% of samples, Giardia intestinalis DNA was detected in 4.5% of samples, Leptospira spp. DNA was detected in 29% of samples, and T. gondii DNA was detected in 31.8% of samples. DNA from three pathogens, C. perfringens, E. histolytica, and T. gondii, were found in residential samples, which accounted for 10 out of 22 samples.
Keywords: water sampling, water-borne pathogen detection, polymerase chain reaction (PCR), gel electrophoresis.
Introduction
The ability of waterborne pathogens to infect a vast population over a relatively short period of time can have profound effects in both developed and developing countries, as demonstrated by past and current epidemics. The World Health Organization asserts about 22% (2.4 million) of 10.8 million deaths in children aged less than five years were caused by diarrheal diseases developed after being exposed to contaminated water sources.[1] The pathogens evaluated in this work are major contributors to diarrheal diseases in developing countries that lack quality drinking water and proper sanitation of sewer systems.
The densely populated areas, limited potable water access, and substandard water sanitation infrastructure of Lima, Peru have contributed to historic epidemics. The 1990s cholera outbreak that claimed the lives of millions of South Americans was attributed to one of the most highly publicized waterborne pathogen outbreaks originating in Peru. A number of waterborne pathogens, including cholera, continue to plague the population of Lima and other underdeveloped areas in Peru. Previous studies in impoverished areas of Peru discovered early childhood infection by gastrointestinal pathogens Giardia intestinalis and Cryptosporidium species are widespread.[2,3,4,5,6] Additionally, other studies have shown the presence of Leptospira species[7], V. cholera and Aeromonas[8,9], Cyclospora cayetanesis[10], and E. coli[11] in a variety of water samples around Peru. Other waterborne pathogens that are common in developing countries, yet have not been identified in Peru waters supplies, include Entamoeboa histolytica[12], Clostridium perfringens[13,14], and Toxoplasma gondii[15,16].Water sanitation and quality has been a concern of previous researchers, yet no study has evaluated both source and delivered water until now.
Past and present day struggles with waterborne pathogens in this region suggest water quality management is substandard or inconsistent. Highly sensitive testing platforms that are cost-effective and produce accurate results in rapid turnaround time can improve diagnostic capacity and relevance. Currently, testing of water samples are conducted by expensive, time-consuming, less sensitive experiments such as immunofluorescence assays and culturing pathogens. Polymerase chain reaction (PCR) is a highly sensitive alternative experiment that detects and amplifies DNA of interest in a shorter period of time. By targeting and replicating DNA, PCR can identify the presence of an organism; and, in some instances, detect specific genes carried by known to lead to disease in humans.[17, 18]
Previous investigators have developed PCR primers to detect the presence of various organisms of interest, due to the importance of gastrointestinal disease identification in medical and food safety industries.[17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27] In this study, we develop a universal protocol derived from published PCR protocols and optimized to decrease material and equipment cost and increase throughput and sensitivity. The creation of such a toolbox is designed to facilitate robust, efficient water monitoring in areas or countries that demonstrate need. Following the characterization of a standardized protocol, we applied our toolbox to water samples from environmental and residential water samples from Lima, Peru to identify the presence or absence of various pathogenic species.
Materials and Methods
Water Sample Acquisition
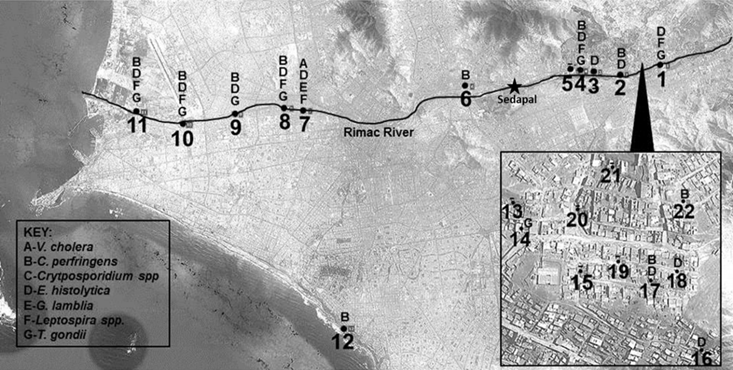
Unlike previous water sampling studies using culture or microscopy methods, relatively few pieces of field and lab equipment were used for this study (Supplemental Table 1). Locations for sampling were determined by satellite imaging along the Rimac River (Figure 1). Peruvian water samples were collected from the ocean shore and 11 locations from the Rimac River, along with 10 residential locations. To collect waters, 1 liter water bailers connected to a 50m retractable line were employed, followed by transferring the bailer volume to 1.5 liter containers. GPS coordinates were recorded at each location by record book, on the collection container, and in the GPS system itself.
Sampling locations and results. This map shows water sampling locations: along the Rimac River which flows westward (1-11), at a beach site (12) and from residential sink taps in region of Ate (13-21). The key identifies each pathogen found at the different sampling locations. The Sedapal drinking water production facility location is noted along the Rimac River, which causes the volume of water in the river to drop significantly due to water capture. As the river continues following west to the Pacific Ocean, wastewater is the primary source of the river's volume.
Water Sample Filtering and Storage
For each sample, 1 liter (as measured by the Steriflip unit) was processed within 24 hours from time of collection. To process the water samples, each sample was filtered using a 25μm polyester filter to remove debris. The flow-through was concentrated onto a 0.22μm membrane (the Stericup's filter) using a handheld vacuum, leaving behind organisms between 25μm and 0.22μm in size (thus excluding viral pathogens). After flow-through, the membrane was simultaneously fixed and disinfected by spraying approximately 0.5mL of 70% ethanol on each membrane and followed by air drying. This last step was an addition to previously published methods, and facilitates disinfection of viable pathogens for safe storage and transport, while also preserving nucleic acids. Filters were then removed with a clean, ethanol-rinsed knife and stored in sterile 50mL Falcon tubes for transport at room temperature.
Positive Controls
To provide template DNA for primer pair characterization in PCR assays, genomic DNA was purchased from ATCC as follows: L. interrogans serovar Copenhageni ATCC-BAA-1198D-5, G. intestinalis Portland-1 ATCC 30888D, V. cholerae MO45 ATCC 51394D-5, V. parahaemolyticus EB101 Y ATCC 17802D-5, C. parvum Iowa strain ATCC PRA-67D. Additional genomic DNA was isolated from ATCC cultures provided by BEI resources as follows: E. histolytica NR 178, C. hominis NR 2520, C. parvum NR 2519, C. perfringens wal-14572 HM 310D, Leptospira interrogans NR 19896, G. lamblia Egypt NR 9231, V. cholera El-tor 34 NR 150, V. cholera O1 NR 147. Toxoplasma gondii DNA was isolated from a maintained laboratory culture of RH∆HXGPRT∆UPRT. DNA isolated from cultured organisms were rapidly thawed from -80oC. The organisms were then centrifuged at 10,000g for 5 minutes, passing through a Qiagen shredder column before DNA isolation using Promega genomic DNA wizard kit per manufacturer instructions. DNA concentration and quality was determined via Nanodrop 2000c. 1ng/µL aliquots of stock concentration DNA were created to minimize contamination risk.
Primer optimization
With known template DNA, 34 primer pairs were screened for amplification specificity and robustness. To identify the best primer pair for each organism, PCR reactions were set up in a 25µL volume as follows: 1X DreamTaq Buffer, 10mM dNTPS, 10µM each forward and reverse primers, 1ng of known DNA (purchased or isolated from sources listed above), 1.25 units of DreamTaq polymerase, and up to 25µL with nuclease-free water. Gradient PCR was used to determine optimal annealing temperatures, and optimized amplification protocols are listed in Table 1. Gel electrophoresis of PCR products was completed using 3% agarose gels containing ethidium bromide with TAE running buffer. Following gel electrophoresis of all runs, the protocol that resulted in a single amplified fragment and/or the stronger band presence was selected for the individual primer pair to be used in sensitivity assays (Table 1).
Primer pair table. Primer pairs with the listed purpose of “Presence / Absence” are designed to detect the presence of listed genus or species by amplifying highly conserved genomic sequences. Primer pairs Chol 14 and Chol 18 detect the presence of the Cholera serovars O139 and O1 respectively, which are known to cause human infection through the presence of the cholera toxin gene (ctxA). The primer pair Chol 18 detects the cholera toxin gene, which is responsible for the diarrheal symptoms characteristic of the cholera infection. The Crp 14 primer pair can detect the two species of Cryptosporidium that cause the most cryptosporidiosis disease in humans, C. parvum and C. hominis. The primer pair can also differentiate between the species by an additional restriction enzyme digestion step with BsiEI on the PCR product (no fragmented bands-C. parvum; two fragments-C. hominis).[22] Other Cryptosporidium species have also been reported to cause cryptosporidiosis in immune competent and compromised individuals, which include C. felis, C. melagridis, C. canis, and C. muris.[6] The “Presence / Absence” primer pair Crp 10 detects the presence of these additional Cryptosporidium species. The Lep 11 primer pair functions to detect the Leptospira-specific virulence factor LipL32, found exclusively in pathogenic leptospires; this allows for differentiation of pathogenic and nonpathogenic Leptospira species.[42]
| Primer ID | Organsim | Sequence (5' to 3') | Amplicon Length | Purpose | Annealing Temperature | Reference |
|---|---|---|---|---|---|---|
| Chol 4 | V. cholerae | TTAAGCSTTTTCRCTGAGAATG (F)AGTCACTTAACCATACAACCCG (R) | 300bp | Presence/Absence | 55°C | 19 |
| Chol 14 | V. cholerae | AGCCTCTTTATTACGGGTGG (F)GTCAAACCCGATCGTAAAGG (R) | 449bp | O139 Serovar | 55°C | 20 |
| Chol 15 | V. cholerae | GTTTCACTGAACAGATGGG (F)GGTCATCTGTAAGTACAAC (R) | 192bp | O1 Serovar | 48.5°C | 20 |
| Chol 18 | V. cholerae | GGTCTTATGCCAGAGGACAG (F)GTTGGGTGCAGTGGCTATAAC (R) | 219bp | Cholera toxin ctxA | 48.5°C | 17 |
| Crp 10 | Cryptosporidium | TCGGTACAGCATCAGGTTCA (F)GTTTTTGCTCCCCCAGTTTT (R) | 368bp | Presence/Absence | 48.5°C | 21 |
| Crp 14 | Cryptosporidium | CAACCCAGAAGTTGAGGTT (F)CTAGTATGCTTCAGACCATGAG (R) | 171/183bp | C. parvum vs. C. hominis | 48.5°C | 22 |
| Clo P 1 | C. perfringens | AAAGATGGCATCATCATTCAAC (F)TACCGTCATTATCTTCCCCAAA (R) | 276bp | Presence/Absence | 48.5°C | 23 |
| Ent 1 | E. histolytica | ATTGTCGTGGCATCCTAACTCA (F)GCGGACGGCTCATTATAACA (R) | 172bp | Presence/Absence | 55°C | 24 |
| Gia 4 | G. lamblia | CAGTACACCTCYGCTCTCGG (F)GTTRTCCTTGCACATCTCC (R) | 410bp | Presence/Absence | 48.5°C | 25 |
| Lep 3 | Leptospira | TAGTGAACGGGATTAGATC (F)GGTCTACTTAATCCGTTAGG (R) | 110bp | Presence/Absence | 48.5°C | 26 |
| Lep 11 | Leptospira | CGCTGAAATGGGAGTTCGTATGATT (F)CCAACAGATGCAACGAAAGATCCTTT (R) | 423bp | Virulence factor LipL32 | 48.5°C | 18 |
| Tox 2 | T. gondii | AGAGACACCGGAATGCGATCT (F)CCCTCTTCTCCACTCTTCAATTCT (R) | 529bp | Presence/Absence | 55°C | 27 |
(F)- forward primer; (R)- reverse primer. Cycling parameters: (1) 90s at 95°C, (2) 40 cycles of 30s at 95°C, 30s at indicated annealing temperature, 60s at 72°C, followed by (3) 10 min 72°C and (4) 4°C.
Sensitivity testing
In order to determine sensitivity of primer pairs using the optimized amplification protocol, isolated DNA for each organism was serially diluted from 100pg/µL to 1 ag/µL using 10-fold dilutions. PCR reactions were set up in 25µL volumes, with DNA concentrations resulting in a range of 1ng/25µL to 10ag/25µL. Negative controls were also performed to control for band formation without template DNA and also amplification of template DNA without primers. All PCR products were run on 3% agarose gels containing ethidium bromide with TAE buffer. Initial detection limits were determined to be the concentration at which the presence of a band was not clearly visible.
To determine the quantity of genomic copies present in each sample following detection limit assays (Table 2), previously determined genome sizes were utilized in the calculation of the genome mass using the average mass of a base pair as 650 Daltons. Genome sizes were used as follows: V. cholerae 4.03MB[28], C. perfringens 3.031MB[29], C. hominis 9.2MB[30], E. histolytica 20MB[31], G. lamblia 11.7MB[32], L. interrogans 4.659MB[33,34], T. gondii 63MB[35]. To further evaluate the sensitivity of these primers in the presence of environmental DNA, local lake water (“environmental background DNA”) was collected and combined with positive control samples to simulate the condition of mixed DNA samples from collection points- see Figure 2.
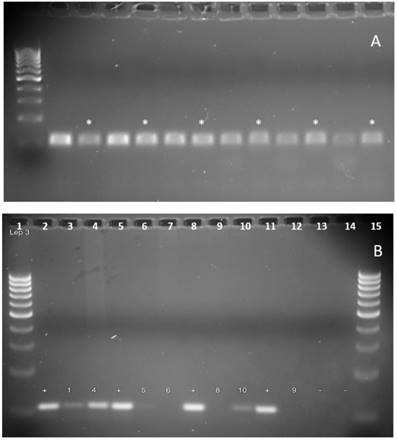
Example gel of Leptospira PCR sensitivity and detection. (A) The gel depicts the sensitivity and specificity of the Leptospira 3 primer pair (F)- TAGTGAACGGGATTAGATAC and (R)- GGTCTACTTAATCCGTTAGG, described previously as primers 16S-P1 and 16S-P2.26 The primer pair predicted amplicon length is 110bp. Leptospira DNA from ATCC (Leptospira interrogans serovar Copenhageni Fiocruz L1-130) was diluted in a series of 10-fold dilutions in sets of two, from 1ng (lanes 2 and 3) to 1ag (lane 12 and 13), by using 1X TE buffer. One sample of each set, marked by '*', contained DNA isolated from a local lake in Omaha, Nebraska not known to contain Leptospira, spiked with 1ng of lab cultured isolated Leptospira DNA. (B) The gel depicts PCR results used to determine presence of Leptospira DNA from collected water samples, using the same Leptospira primer pair as above. '+' indicates the positive control (known DNA obtained from ATCC), '-' indicates negative control (primer pair only, no template DNA), and an annealing temperature of 48.5°C was used for used for the PCR cycle. Lanes 1 and 15 contain Fisher exACTgene 100bp ladder. The numbers correspond to samples numbers in Table 2. Presence of Leptospira was identified in samples 1, 4, 5, and 10.
Primer pair minimum detection limits. Primer sensitivity listed by the minimum quantity of genomic copies required for detection of the specified DNA fragment. Bacteria sampled here are generally considered to maintain a single genome copy (V. cholerae, C. perfringens, and Leptospira), whereas the sampled parasites vary in genome count (typically 2-4 copies). Tests were performed using 10-fold dilutions of template DNA with a starting concentration of 1ng per reaction and were measured either from DNA isolated from lab cultured organisms, or intact organisms spiked into an environmental sample consisting of Nebraska lake water (which was negative for all tested organisms). Minimum tested template concentrations varied by organism between 10 fg to 10 ag per reaction. The concentration of nonspecific DNA, isolated from an environmental sample, was maintained at 1ng per reaction. No bands were present in negative controls.
| Primer ID | Organsim | Purpose | Minimum detection limit (Genomic copies) Lab cultured sample | Minimum detection limit (Genomic copies) Environmental sample-spiked |
|---|---|---|---|---|
| Chol 4 | V. cholerae | Presence/Absence | 230 | 230 |
| Chol 14 | V. cholerae | O139 Serovar | 2300 | 2300 |
| Chol 15 | V. cholerae | O1 Serovar | 230 | 230 |
| Chol 18 | V. cholerae | Cholera toxin ctxA | 23 | 2300 |
| Crp 10 | Cryptosporidium | Presence/Absence | 3 | 3 |
| Crp 14 | Cryptosporidium | C. parvum vs. C. hominis | 1 | 1 |
| Clo P 1 | C. perfringens | Presence/Absence | 101 | 101 |
| Ent 1 | E. histolytica | Presence/Absence | 5 | 5 |
| Gia 4 | G. lamblia | Presence/Absence | 79 | 79 |
| Lep 3 | Leptospira | Presence/Absence | 199 | 199 |
| Lep 11 | Leptospira | Virulence factor LipL32 | 199 | 19 |
| Tox 2 | T. gondii | Presence/Absence | 14 | 14 |
Pathogen Detection
Upon returning to the laboratory (approximately 1 month after acquisition & fixation for all samples), filters were soaked in 2.5mL TE Buffer pH7.4 for 24hr. After soaking, the filters were scraped with a cell scraper, and the suspension was passed through a QiaShredder column. The filter was then cut into <5mmx5mm pieces and resuspended in 2mL TE buffer. The volume, including filter pieces, was vortexed vigorously then spun down at 2000rpm for 10 minutes. The supernatant was passed through a QiaShredder column and combined with the previous column flow-through. All flow-through was spun a final time at 13000rpm. The supernatant was removed, leaving a pellet behind for processing using a Promega Wizard DNA isolation kit according to the manufacturer protocol.
Isolated DNA concentration and quality was confirmed using a Nanodrop 2000c. Supplemental Table 2 shows the sample location and the quantity and quality DNA isolated from each Peruvian water sample. After determining concentration and purity of the isolated DNA, samples were stored at -20°C until further use. PCR reactions were subsequently run on each DNA sample, screening for the presence or absence of an organism or pathogen-related gene by specific PCR primers. Thermo Scientific DreamTaq PCR kit was used for all reactions in a total volume of 25µL. Cycling parameters were run as follows: (1) 90s at 95°C, (2) 40 cycles of 30s at 95°C, 30s at indicated annealing temperature listed in the figure legends, 60s at 72°C, followed by (3) 10 min 72°C and (4) 4°C. Both positive and negative controls were run in conjunction with the water sample DNA. Positive controls were run with organisms or DNA supplied by ATCC; and two negative controls were run to identify non-specific amplification: 1) reactions lacking any DNA template and 2) reactions lacking primer pairs. Results from the water sample PCRs were run against the positive and negative controls on 3% agarose gels containing ethidium bromide with TAE running buffer. Bands were visualized on a UV box to determine any amplification (Figure 2).
Results
Primer Pair Selection and Sensitivity
Following initial test amplifications, minimum detection limit assays were performed on 34 primers pairs to determine the sensitivity of the primer pair to a known target. After showing the visualization of bands decreasing as DNA concentration decreased, select primers were further tested to amplify target DNA in the presence of foreign DNA. The primer pairs that most consistently resulted in robust amplification of intended products are shown in table 1. In order to quantify our results, the minimum detection limits in units of genomic copies are shown in table 2. No non-specific amplification was shown in the PCRs containing foreign DNA, suggesting high specificity of the selected primers was sustained.
Water Sample Testing
The Peru water samples, 22 total, were screened using 12 different pathogen-detecting assays. Table 3 shows all 22 samples and the pathogen presence or absence. In the 22 samples test: cholera toxin was detected in 4.5% of samples, C. perfringens was detected in 50% of samples, E. histolytica was detected in 54.5% of samples, Giardia intestinalis was detected in 4.5% of samples, Leptospira spp. was detected in 29% of samples, and T. gondii was detected in 31.8% of samples. Most of the pathogens found were detected in the samples from the Rimac River. Only C. perfringens, E. histolytica, and T. gondii were found in the residential samples, suggesting that water treated by the Sedapal drinking water facility is mostly efficient at removing waterborne pathogens. Figure 2 depicts an example agarose gel.
Results from sampling locations. Table 3 demonstrates the detection results from the water sampling of 22 locations in Lima, Peru. Sample IDs reflect the corresponding locations in Figure 1. “+” indicates detection of presence, specific species or virulence factor of pathogens, whereas “-“ indicates no presence found. In the 22 water samples tested, cholera toxin was detected in 4.5% of samples, C. perfringens was detected in 50% of samples, E. histolytica was detected in 54.5% of samples, G. intestinalis was detected in 4.5% of samples, Leptospira spp. was detected in 29% of samples, and T. gondii was detected in 31.8% of samples. “**” indicates insufficient remaining sample volume for retesting.
| Sample ID | Chol 4 (+/-) | Chol 14 (O1 serovar +/- ) | Chol 15 (O139 serovar +/- ) | Chol 18 (Cholera toxin ctxA +/- ) | Clo P 1 (+/-) | Crp 10 (+/-) | Crp 14 (C. parvum/C. hominis) | Ent 1 (+/-) | Gia 4 (+/-) | Lep 3 (+/-) | Lep 11 (Virulence factor LipL32) | Tox 2 (+/-) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| River 1 | - | - | - | - | - | - | - | + | - | + | - | + |
| River 2 | - | - | - | - | + | - | - | + | - | ** | - | - |
| River 3 | - | - | ** | - | - | - | - | + | - | - | - | - |
| River 4 | - | - | - | - | + | - | - | + | - | + | - | + |
| River 5 | - | - | - | - | - | - | - | + | - | - | - | - |
| River 6 | - | - | - | - | + | - | - | - | - | - | - | - |
| River 7 | - | - | - | + | - | - | - | + | + | - | - | |
| River 8 | - | - | - | - | + | - | - | + | - | + | - | + |
| River 9 | - | - | - | - | + | - | - | + | - | - | - | + |
| River 10 | - | - | - | - | + | - | - | + | - | + | - | + |
| River 11 | - | - | - | - | + | - | - | + | - | + | - | + |
| Ocean 12 | - | - | - | + | - | - | - | - | - | - | - | |
| Residential 13 | - | - | - | - | - | - | - | - | - | - | - | - |
| Residential 14 | - | - | - | - | - | - | - | - | - | - | - | + |
| Residential 15 | - | - | - | - | - | - | - | - | - | - | - | - |
| Residential 16 | - | - | - | - | - | - | - | + | - | - | - | - |
| Residential 17 | - | - | - | - | + | - | - | + | - | - | - | - |
| Residential 18 | - | - | - | - | - | - | - | + | - | - | - | - |
| Residential 19 | - | - | - | - | - | - | - | - | - | - | - | - |
| Residential 20 | - | - | - | - | - | - | - | - | - | - | - | - |
| Residential 21 | - | - | - | - | - | - | - | - | - | - | - | - |
| Residential 22 | - | - | - | - | + | - | - | - | - | - | - | - |
| Total | 0/22 | 0/22 | 0/21 | 1/22 | 11/22 | 0/22 | 0/22 | 12/22 | 1/22 | 6/21 | 0/22 | 7/22 |
Discussion
Some current detection methods for waterborne pathogens, i.e. culture and microscopy, are manageable, yet not feasible to be completed in a timely, cost-efficient manner. PCR can be an effective alternative that has improved sensitivity and can be done in a short period of time for less money. Though there can be drawbacks to PCR, such as non-specific amplification or detection of nonviable organisms, our study shows the ability of DNA isolation protocol and PCR assay to survey environmental and residential water samples in the presence of background DNA.
Using previously established primer pairs, PCR assays were run to determine an optimized set of primers and amplification protocols by screening PCR for robust amplification using known DNA sources. After the best primer pairs were identified, the sensitivity of each pair was tested using 10-fold dilutions of known template DNA in the presence of environmental background DNA. In 11 of 12 PCR assays, no detectable inhibition was found. The Chol 18 primer pair, which detect the presence of cholera toxin ctxA, decreased sensitivity 100-fold in the presence of environmental background DNA.[36]
Previous studies describe the prevalence of disease cause by Leptospira, Cryptosporidium, Cholera, and Giardia species in Peru[2,3,4,5,6,7,9,25,37,38]; however, few studies have focused on the quality of water available to those with and without access to potable water connections. Sample 11, the westernmost sample collected from the Rimac River and closer to the mountain water source, contained six out of the seven pathogens discussed in this study: V. choleraae, Giardia intestinalis, Clostridium perfringens, Entamoeba histolytica, Leptospira spp., and Toxoplasma gondii (Table 3). In contrast, sample 12, collected in the Miraflores district near the Pacific Ocean, only had the detectable presence of C. perfringens- a limited result likely due to dilution.
Residential samples collected in the Ate region of Lima, Peru showed low frequency of contamination from pathogenic organisms. This finding can be attributed to the chlorination of water at the drinking water facility, Sedapal. Samples collected at residential locations 13, 15, 19, 20, and 21 did not contain detectable quantities of DNA. Samples 16, 17, and 18 were determined to contain C. perfringens; and sample 14 tested positive for T. gondii. The low number of detectable pathogens suggests that contamination is highly localized. This confirms previous publications that cite examples of localized contamination due to a number of factors such as biofilm, non-continuous pressure, and leaks. [39, 40, 41]
Conclusion
The importance of a highly sensitive, cost-effective, timely test kit for waterborne pathogens is paramount for screening a number of water supplies in both developed and undeveloped countries. While current testing, i.e. culture methods and microscopy, are useful, this study describes the DNA isolation and PCR methods that can be utilized to test a large number of water samples with quick turnaround and accuracy. The methods discussed in this study led to an identification of several pathogenic organisms in Peruvian waters along the Rimac River and in the Ate region of Lima.
Future studies and testing of water supplies can be expanded to include a number of waterborne pathogens, including Cyclospora species, pathogenic E. coli, Salmonella typhi, and Hepatitis A and E viruses. Additionally, sampling volumes can be increased to reflect an average daily intake of water in the areas of water sampling. By increasing the volume of samples taken, a more accurate picture of pathogenic species can be seen. The seriousness of waterborne pathogen contamination in worldwide water supplies cannot be understated. A total of 780 million people do not have access to clean water, and exposure to contaminated water sources can result in death.42 By utilizing a simple water testing method that is cheap, highly sensitive, and timely, drastic improvements can be made to water quality worldwide.
Supplementary Material
Supplementary tables.
Acknowledgements
This work was supported by the NIH National Institute for General Medical Science (NIGMS) (P20GM103427). Further support was provided by The University of Nebraska DNA Sequencing Core, which receives support NIH COBRE P30GM110768 as well as The Fred & Pamela Buffett Cancer Center Support Grant P30CA036727. The GRACA and FUSE programs of the University of Nebraska at Omaha additionally supported this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Forsberg BC, Petzold MG, Tomson G, Allebeck P. Diarrhoea Case management in low- and middle-income countries—an unfinished agenda. Bulletin of World Health Organization. 2007:85 (1)
2. Hollm-Delgado MG, Gilman RH, Bern C, Cabrera L, Sterling CR, Black RE, Checkley W. Lack of an adverse effect of Giardia intestinalis infection on the health of Peruvian children. Am J Epidemiol. 2008;168(6):647-55
3. Cooper MA, Sterling CR, Gilman RH, Cama V, Ortega Y, Adam RD. Molecular analysis of household transmission of Giardia lamblia in a region of high endemicity in Peru. J Infect Dis. 2010;202(11):1713-21
4. Cama VA, Bern C, Roberts J, Cabrera L, Sterling CR, Ortega Y, Gilman RH, Xiao L. Cryptosporidium species and subtypes and clinical manifestations in children, Peru. Emerg Infect Dis. 2008;14(10):1567-74
5. Nundy S, Gilman RH, Xiao L, Cabrera L, Cama R, Ortega YR, Kahn G, Cama VA. Wealth and its associations with enteric parasitic infections in a low-income community in Peru: use of principal component analysis. Am J Trop Med Hyg. 2011;84(1):38-42
6. Xiao L, Bern C, Limor J, Sulaiman I, Roberts J, Checkley W, Cabrera L, Gilman RH, Lal AA. Identification of 5 types of Cryptosporidium parasites in children in Lima, Peru. J Infect Dis. 2001;183(3):492-7
7. Segura ER, Ganoza CA, Campos K, Ricaldi JN, Torres S, Silva H, Céspedes MJ, Matthias MA, Swancutt MA, López Liñán R, Gotuzzo E, Guerra H, Gilman RH, Vinetz JM; Peru-United States Leptospirosis Consortium. Clinical spectrum of pulmonary involvement in leptospirosis in a region of endemicity, with quantification of leptospiral burden. Clin Infect Dis. 2005;40(3):343-51
8. Gilman RH, Marquis GS, Ventura G, Campos M, Spira W, Diaz F. Water cost and availability: key determinants of family hygiene in a Peruvian shantytown. Am J Public Health. 1993;83(11):1554-8
9. Lipp EK, Rivera IN, Gil AI, Espeland EM, Choopun N, Louis VR, Russek-Cohen E, Huq A, Colwell RR3. Direct detection of Vibrio cholerae and ctxA in Peruvian coastal water and plankton by PCR. Appl Environ Microbiol. 2006;9(6):3676-80
10. Sturbaum GD, Ortega YR, Gilman RH, Sterling CR, Cabrera L, Klein DA. Detection of Cyclospora cayetanensis in wastewater. Appl Environ Microbiol. 1998;64(6):2284-6
11. Oswald WE, Lescano AG, Bern C, Calderon MM, Cabrera L, Gilman RH. Fecal contamination of drinking water within peri-urban households, Lima, Peru. Am J Trop Med Hyg. 2007;77(4):699-704
12. Stark D, van Hal S, Fotedar R, Butcher A, Marriott D, Ellis J, Harkness J. Comparison of stool antigen detection kits to PCR for diagnosis of amebiasis. J Clin Microbiol. 2008;46(5):1678-81
13. Ahmed W, Huygens F, Goonetilleke A, Gardner T. Real-time PCR detection of pathogenic microorganisms in roof-harvested rainwater in Southeast Queensland, Australia. Appl Environ Microbiol. 2008;74(17):5490-6
14. Manafi M, Waldherr K, Kundi M. Evaluation of CP Chromo Select Agar for the enumeration of Clostridium perfringens from water. Int J Food Microbiol. 2013;167(1):92-5
15. Hill D, Dubey JP. Toxoplasma gondii: transmission, diagnosis and prevention. Clin Microbiol Infect. 2002;8(10):634-40
16. Villena I, Aubert D, Gomis P, Ferté H, Inglard JC, Denis-Bisiaux H, Dondon JM, Pisano E, Ortis N, Pinon JM. Evaluation of a strategy for Toxoplasma gondii oocyst detection in water. Appl Environ Microbiol. 2004;70(7):4035-9
17. Mehrabadi JF, Morsali P, Nejad HR, Imani Fooladi AA. Detection of toxigenic Vibrio cholerae with new multiplex PCR. J Infect Public Health. 2012;5(3):263-7
18. Levett PN, Morey RE, Galloway RL, Turner DE, Steigerwalt AG, Mayer LW. Detection of pathogenic leptospires by real-time quantitative PCR. J Med Microbiol. 2005;54(Pt 1):45-9
19. Colwell RR, Seidler RJ, Kaper J, Joseph SW, Garges S, Lockman H, Maneval D, Bradford H, Roberts N, Remmers E, Huq I, Huq A. Occurrence of Vibrio cholerae serotype O1 in Maryland and Louisiana estuaries. Appl Environ Microbiol. 1981;41(2):555-8
20. Hoshino K, Yamasaki S, Mukhopadhyay AK, Chakraborty S, Basu A, Bhattacharya SK, Nair GB, Shimada T, Takeda Y. Development and evaluation of a multiplex PCR assay for rapid detection of toxigenic Vibrio cholerae O1 and O139. FEMS Immunol Med Microbiol. 1998;20(3):201-7
21. Bouzid M, Tyler KM, Christen R, Chalmers RM, Elwin K, Hunter PR. Multi-locus analysis of human infective Cryptosporidium species and subtypes using ten novel genetic loci. BMC Microbiol. 2010;10:213
22. Lee SH, Joung M, Yoon S, Choi K, Park WY, Yu JR. Multiplex PCR Detection of Waterborne Intestinal Protozoa: Microsporidia, Cyclospora, and Cryptosporidium. Korean J Parasitol. 2010;48(4):297-301
23. Yoo HS, Lee SU, Park KY, Park YH. Molecular typing and epidemiological survey of prevalence of Clostridium perfringens types by multiplex PCR. J Clin Microbiol. 1997;35(1):228-32
24. Verweij JJ, Blangé RA, Templeton K, Schinkel J, Brienen EA, van Rooyen MA, van Lieshout L, Polderman AM. Simultaneous detection of Entamoeba histolytica, Giardia lamblia, and Cryptosporidium parvum in fecal samples by using multiplex real-time PCR. J Clin Microbiol. 2004;42(3):1220-3
25. Peréz Cordon G, Cordova Paz Soldan O, Vargas Vasquez F, Velasco Soto JR, Sempere Bordes Ll, Sanchez Moreno M, Rosales MJ. Prevalence of enteroparasites and genotyping of Giardia lamblia in Peruvian children. Parasitol Res. 2008;103:459-465
26. Bedir O, Kilic A, Atabek E, Kuskucu AM, Turhan V, Basustaoglu AC. Simultaneous detection and differentiation of pathogenic and nonpathogenic Leptospira spp. by multiplex real-time PCR (TaqMan) assay. Pol J Microbiol. 2010;59(3):167-73
27. Lélu M, Villena I, Dardé ML, Aubert D, Geers R, Dupuis E, Marnef F, Poulle ML, Gotteland C, Dumètre A, Gilot-Fromont E. Quantitative estimation of the viability of Toxoplasma gondii oocysts in soil. Appl Environ Microbiol. 2012;78(15):5127-32
28. Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000;406(6795):477-83
29. Shimizu T, Ohtani K, Hirakawa H, Ohshima K, Yamashita A, Shiba T, Ogasawara N, Hattori M, Kuhara S, Hayashi H. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc Natl Acad Sci U S A. 2002;99(2):996-1001
30. Xu P, Widmer G, Wang Y, Ozaki LS, Alves JM, Serrano MG, Puiu D, Manque P, Akiyoshi D, Mackey AJ, Pearson WR, Dear PH, Bankier AT, Peterson DL, Abrahamsen MS, Kapur V, Tzipori S, Buck GA. The genome of Cryptosporidium hominis. Nature 431. 2004(7012):1107-12
31. Lorenzi HA, Puiu D, Miller JR, Brinkac LM, Amedeo P, Hall N, Caler EV. New assembly, reannotation and analysis of the Entamoeba histolytica genome reveal new genomic features and protein content information. PLoS Negl Trop Dis. 2010;4(6):e716
32. Morrison HG, McArthur AG, Gillin FD, Aley SB, Adam RD, Olsen GJ, Best AA, Cande WZ, Chen F, Cipriano MJ, Davids BJ, Dawson SC, Elmendorf HG, Hehl AB, Holder ME, Huse SM, Kim UU, Lasek-Nesselquist E, Manning G, Nigam A, Nixon JE, Palm D, Passamaneck NE, Prabhu A, Reich CI, Reiner DS, Samuelson J, Svard SG, Sogin ML. Genomic minimalism in the early diverging intestinal parasite Giardia lamblia. Science 317. 2007(5846):1921-6
33. Nascimento AL, Ko AI, Martins EA, Monteiro-Vitorello CB, Ho PL, Haake DA, Verjovski-Almeida S, Hartskeerl RA, Marques MV, Oliveira MC, Menck CF, Leite LC, Carrer H, Coutinho LL, Degrave WM, Dellagostin OA, El-Dorry H, Ferro ES, Ferro MI, Furlan LR, Gamberini M, Giglioti EA, Góes-Neto A, Goldman GH, Goldman MH, Harakava R, Jerônimo SM, Junqueira-de-Azevedo IL, Kimura ET, Kuramae EE, Lemos EG, Lemos MV, Marino CL, Nunes LR, de Oliveira RC, Pereira GG, Reis MS, Schriefer A, Siqueira WJ, Sommer P, Tsai SM, Simpson AJ, Ferro JA, Camargo LE, Kitajima JP, Setubal JC, Van Sluys MA. Comparative genomics of two Leptospira interrogans serovars reveals novel insights into physiology and pathogenesis. J Bacteriol. 2004;186(7):2164-2172
34. Ren SX, Fu G, Jiang XG, Zeng R, Miao YG, Xu H, Zhang YX, Xiong H, Lu G, Lu LF, Jiang HQ, Jia J, Tu YF, Jiang JX, Gu WY, Zhang YQ, Cai Z, Sheng HH, Yin HF, Zhang Y, Zhu GF, Wan M, Huang HL, Qian Z, Wang SY, Ma W, Yao ZJ, Shen Y, Qiang BQ, Xia QC, Guo XK, Danchin A, Saint Girons I, Somerville RL, Wen YM, Shi MH, Chen Z, Xu JG, Zhao GP. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature 422. 2003(6934):888-893
35. Gajria B, Bahl A, Brestelli J, Dommer J, Fischer S, Gao X, Heiges M, Iodice J, Kissinger JC, Mackey AJ, Pinney DF, Roos DS, Stoeckert CJ Jr, Wang H, Brunk BP. ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res. 2008;36(Database issue):D553-6
36. Mekalanos JJ. Duplication and amplification of toxin genes in Vibrio cholerae. Cell 35. 1983(1):253-63
37. Ganoza CA, Matthias MA, Collins-Richards D, Brouwer KC, Cunningham CB, Segura ER, Gilman RH, Gotuzzo E, Vinetz JM. Determining risk for severe leptospirosis by molecular analysis of environmental surface waters for pathogenic Leptospira. PLoS Med. 2006;3(8):e308
38. Ganoza CA, Matthias MA, Saito M, Cespedes M, Gotuzzo E, Vinetz JM. Asymptomatic renal colonization of humans in the peruvian Amazon by Leptospira. PLoS Negl Trop Dis. 2010;4(2):e612
39. Cho EJ, Yang JY, Lee ES, Kim SC, Cha SY, Kim ST, Lee MH, Han SH, Park YS. A Waterborne Outbreak and Detection of Cryptosporidium Oocysts in Drinking Water of an Older High-Rise Apartment Complex in Seoul. Korean J Parasitol. 2013;51(4):461-466
40. Lazcano CA. Failures and problems of urban disinfection. CEPIS; OPS. Regional symposium on water quality: effective disinfection. Lima, CEPIS. 1998:1-11
41. Kumpel E, Nelson KL. Comparing microbial water quality in an intermittent and continuous piped water supply. Water Res. 2013;47(14):5176-88
42. Haake DA, Chao G, Zuerner RL, Barnett JK, Barnett D, Mazel M, Matsunaga J, Levett PN, Bolin CA. The leptospiral major outer membrane protein LipL32 is a lipoprotein expressed during mammalian infection. Infect Immun. 2000;68(4):2276-85
Author contact
![]() Corresponding author: Paul H. Davis, Department of Biology, University of Nebraska at Omaha, 6001 Dodge St. Omaha, Nebraska 68182. Email: pdavisedu; Phone: 402-554-3379
Corresponding author: Paul H. Davis, Department of Biology, University of Nebraska at Omaha, 6001 Dodge St. Omaha, Nebraska 68182. Email: pdavisedu; Phone: 402-554-3379