ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2017; 5:16-31. doi:10.7150/jgen.18856 This volume Cite
Research Paper
Exploration of small RNA-seq data for small non-coding RNAs in Human Colorectal Cancer
Srinivas V Koduru1 ![]() , Amit K Tiwari2, Sprague W Hazard3, Milind Mahajan4, Dino J Ravnic1
, Amit K Tiwari2, Sprague W Hazard3, Milind Mahajan4, Dino J Ravnic1
1. Division of Plastic Surgery, Department of Surgery, College of Medicine, Pennsylvania State University, Hershey, PA, USA;
2. Department of Pharmacology & Experimental Therapeutics, College of Pharmacy & Pharmaceutical Sciences, University of Toledo - Health Sciences Campus, Toledo, OH, USA;
3. Department of Anesthesia, College of Medicine, Pennsylvania State University, Hershey, PA, USA;
4. Genomics Facility, Department of Genetics and Genomics Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Published 2017-2-28
Abstract

Background: Improved healthcare and recent breakthroughs in technology have substantially reduced cancer mortality rates worldwide. Recent advancements in next-generation sequencing (NGS) have allowed genomic analysis of the human transcriptome. Now, using NGS we can further look into small non-coding regions of RNAs (sncRNAs) such as microRNAs (miRNAs), Piwi-interacting-RNAs (piRNAs), long non-coding RNAs (lncRNAs), and small nuclear/nucleolar RNAs (sn/snoRNAs) among others. Recent studies looking at sncRNAs indicate their role in important biological processes such as cancer progression and predict their role as biomarkers for disease diagnosis, prognosis, and therapy. Results: In the present study, we data mined publically available small RNA sequencing data from colorectal tissue samples of eight matched patients (benign, tumor, and metastasis) and remapped the data for various small RNA annotations. We identified aberrant expression of 13 miRNAs in tumor and metastasis specimens [tumor vs benign group (19 miRNAs) and metastasis vs benign group (38 miRNAs)] of which five were upregulated, and eight were downregulated, during disease progression. Pathway analysis of aberrantly expressed miRNAs showed that the majority of miRNAs involved in colon cancer were also involved in other cancers. Analysis of piRNAs revealed six to be over-expressed in the tumor vs benign cohort and 24 in the metastasis vs benign group. Only two piRNAs were shared between the two cohorts. Examining other types of small RNAs [sn/snoRNAs, mt_rRNA, miscRNA, nonsense mediated decay (NMD), and rRNAs] identified 15 sncRNAs in the tumor vs benign group and 104 in the metastasis vs benign group, with only four others being commonly expressed. Conclusion: In summary, our comprehensive analysis on publicly available small RNA-seq data identified multiple differentially expressed sncRNAs during colorectal cancer progression at different stages compared to normal colon tissue. We speculate that deciphering and validating the roles of sncRNAs may prove useful in colorectal cancer prognosis, diagnosis, and therapy.
Keywords: Bioinformatics, colorectal cancer, next-generation genomic sequencing, small non-coding RNA, sncRNA, miRNA, snoRNA.
Introduction
Colorectal cancer (CRC) is the third-most common cancer diagnosed in the United States in both men and woman, and the American Cancer Society estimates 134,490 new cases and 49,190 deaths in 2016 [1]. Despite recent improvements in cancer care and awareness, incidences are still steadily growing. Although colonoscopy has allowed for earlier detection, a better understanding of tumor biology and prognosis are still needed. Recently, advances in RNA sequencing have increased the understanding of the molecular mechanisms behind CRC. Changes in whole transcriptome profiles seem to underlie specific phenotypes. Small non-coding RNAs consist of 17-120 nucleotides, in their mature form [2] and play crucial roles in the development and progression of cancer. Innovative technologies have allowed researchers to focus on the role of small RNAs in disease progression which may contribute to the development of novel therapeutic targets. An increasing number of published studies have focused on miRNAs (17-22 nucleotides) [3], piRNAs (26-33 nucleotides) [4], small nucleolar RNAs (70-120 nucleotides) [5], and lncRNAs (>200 nucleotides) [6]. Using whole genome sequencing, present studies aim to understand the functions and transcriptional regulation of miRNA genes. Piwi-interacting RNAs (piRNAs) are one of the largest classes of the small non-coding RNAs family, and have been implicated in DNA methylation and post transcriptional-mediated repression of transposable elements [7]. However, other hypothesized functions of piRNAs remain largely unexplored [8]. Similarly, limited functional information is available for long non-coding RNAs (lncRNAs) [9]. Likewise, small nuclear RNAs (snRNAs) in eukaryotes consist of small nucleolar RNAs (snoRNAs), which carry out the fundamental role of ribosomal (rRNA) and transfer RNA (tRNA) modification and processing [10]. Two classes of snoRNAs, C/D and box H/ACA snoRNAs, have been shown to differ in sequence and structure [10].
In the present study, we carried out in-depth data analysis to identify the transcriptomic expression “signatures” of small RNAs from benign, tumor, and metastatic tissue samples from colorectal cancer patients, using existing datasets. We thoroughly analyzed the most predominant ncRNA expression in each sample type along with signaling pathways for possible biomarker development and identification of therapeutic targets in colorectal cancer.
Materials and Methods
Samples and data assembly
Colorectal cancer (CRC) small RNA sequencing (eight matched patients; 8 benign, 8 tumors, and 8 metastasis) datasets (project no: PRJNA201245; GEO: GSE46622) were downloaded from the NIH bioproject [11]. Detailed clinical information of patients was previously mentioned by Rohr et al [11]. Raw files were downloaded as sequence raw archive (SRA) files that were converted to FASTQ using SRA tool Kit version 2.5.7. Data was assembled using PartekFlow® software, version 5.0 (Partek, Inc., St. Louis, MO, USA). Converted FASTQ files were uploaded to the PartekFlow® server and remapped to human genome hg19 (UCSC Genome Browser). Transcript abundances were determined and expression values were represented using reads per kilobases of exon model per million mapped reads (RPKM). All small RNA expression RPKM values >1 and representing at least 10% of the samples were considered robustly expressed and used for further analysis. Expression matrices were aligned to clinical pathologic features in order to compare miRNA, piRNA, lncRNA, and snRNA levels for association with specific CRC phenotypes. Statistical analysis between benign, tumor, and metastasis groups were carried out using the non-parametric Mann-Whitney U test followed by false discovery rate (FDR) correction through the Benjamin-Hochberg method, with the default p value <0.05 considered statistically significant [12].
Assembly of miRNA, piRNA, lncRNA and snoRNA annotations
Small RNA sequencing data was trimmed and aligned to the whole human genome (hg19), and BWA-0.7.12 aligner (BWA-MEM) with a few modifications (mismatch penalty 2, gap open penalty 6, clipping penalty 4 and alignment score cutoff 15) for short read mapping [13]. miRNAs were annotated from miRBase version 21 (http://www.mirbase.org/), which contains more than 1900 high confidence miRNAs [14]. piRNA data was generated and annotated from piRBase (regulatoryrna.org/database/piRNA), which is manually curated with a focus on piRNA functional analysis [15]. lncRNAs were quantified using reference annotation LNCipedia (http://www.lncipedia.org) version 3.1, downloaded from all coordinates relative to the hg19 reference genome [16]. Total small RNA (including miRNA, piRNA, snRNA, mtRNA, snoRNA, piRNA, tRF3, tRF5, tRNA, rRNA) was annotated following download of GenCode version 21 (www.GenCodegenes.org), which provides comprehensive, up-to-date information on human small non-coding RNAs, with specific regard to small nuclear and nucleolar RNAs.
Biological processes and gene network visualization: MetaCore
To investigate biological pathway interactions of small RNA expression, we used MetaCore pathway analysis of differentially expressed genes (Thomson Reuters, New York, NY) [13, 17], to delineate functional gene networks. This was based on differentially regulated gene lists with p<0.05 from each group (tumor vs benign, metastasis vs benign, metastasis vs tumor) as input (Data analyzed by Gene Arrays, New York, USA) to generate disease biomarkers and GO terms.
Statistical Analysis
All data sets were subjected to paired student's t test with “p” value <0.05 being considered statistically significant. Furthermore, the Benjamini and Hochberg multiple testing adjustment method was applied to all small RNA sequencing studies and pathway analysis.
Results and Discussion
Analysis of miRNA sequencing of colorectal cancer tissue samples
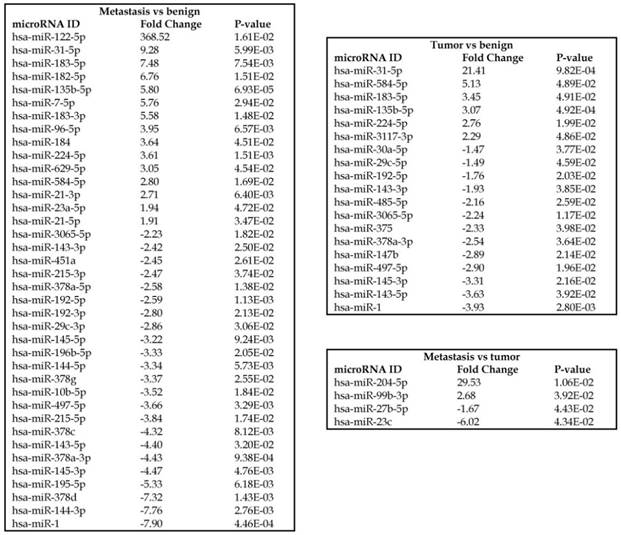
To identify miRNA differential expression in colorectal cancer patient-matched tissue samples, data was downloaded from the GEO series accession number GSE46622. Using PartekFlow software miRNA analysis, we identified 19 miRNAs in the tumor vs benign cohort, and 38 miRNAs in the metastasis vs benign cohort, that were statistically significant. Top upregulated/downregulated miRNAs were determined in both tumor, metastasis stages (Figures 1A and 1B, Table 1), and hierarchical clustering/Venn diagram was compared for each group (Figure 1C). Our findings were very similar to those originally published by Rohr et al [11], in which one of the top downregulated microRNAs was miR-1. It is downregulated during disease progression from tumor (4-fold) to metastasis (8-fold, Figure 1B), along with miR-145, miR-378a, miR-143 and miR-497 commonly regulated in both tumor and metastasis samples. Additionally, miR-31, miR-183, miR-135b, miR224 and miR-584 were upregulated with disease progression (Figure 1A, 1B and 1C). All of the miRNAs with fold change were listed in Table 1. miR-1 downregulation is consistent with other studies that showed its downregulation in colorectal cancers [18]. Several other miRNAs were expressed at low levels in normal tissue, strongly upregulated in tumors, and continued to be further upregulated in late stage (metastatic) CRC. miR-31 (9.3-fold), miR-183 (7.5-fold), miR-135b (6-fold) and miR-224 (3.6-fold), were all significantly expressed in metastasis in the sequencing data, in agreement with other studies demonstrating the same expression pattern in CRC and other disease states [19-21]. Specifically, miR-31 expression in benign samples was negligible and increased significantly with disease progression, this data is similar to previous reports showing increased presence of miR-31 in several cancer types with metastasis [22].
Differential expression of miRNAs in tumor and metastasis of colorectal cancer specimens: Top four statistically upregulated (Figure 1A) and downregulated (Figure 1B) miRNAs and dendrogram view of all miRNAs that are statistically significant (p≤0.05) in the tumor and metastatic tissue samples and associated Venn diagram (Figure 1C).
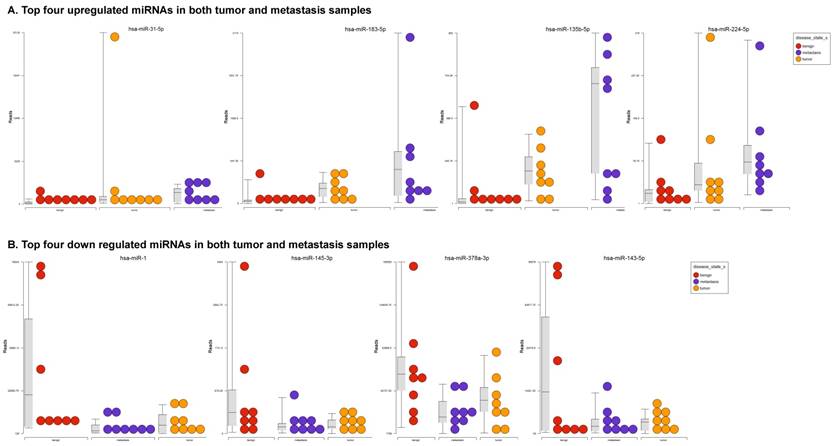
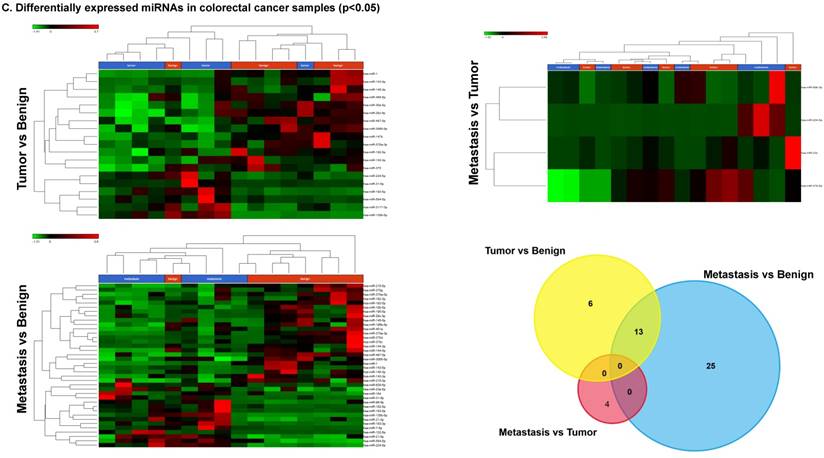
Differential expression of miRNAs in three groups.
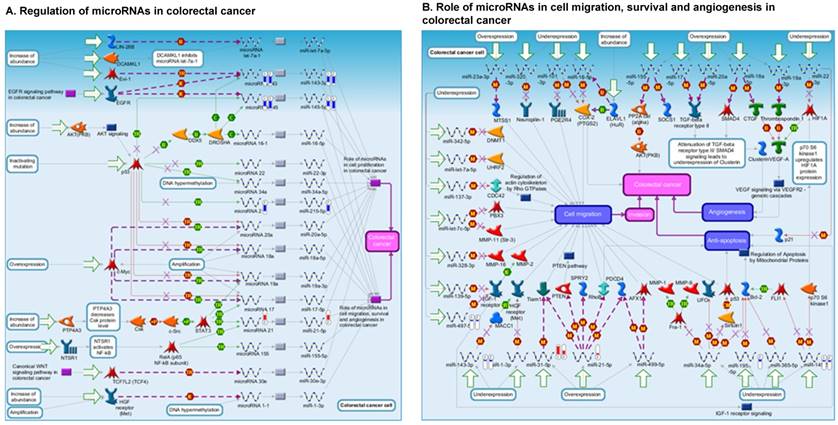
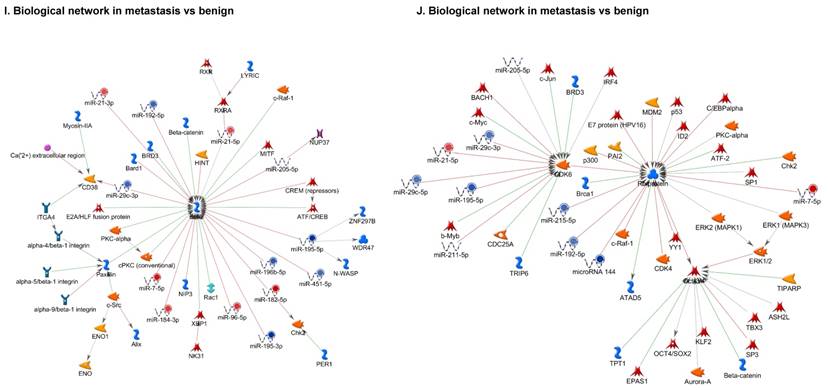
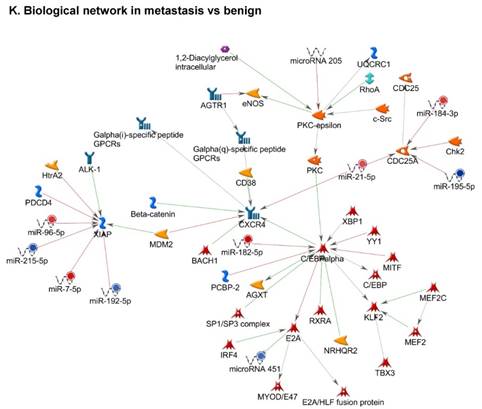
Once miRNA gene expression was determined, we performed biological pathway analysis, using MetaCore software (previous known as GeneGo; v6.25), to identify candidate “molecular signatures.” Pathway analysis on differentially expressed benign, tumor, and metastasis samples were obtained. As expected, most of the affected miRNAs were involved in cell regulation, cell proliferation, migration, and angiogenesis of colorectal cancer (Figure 2A). Gene ontology (Figure 2B) and disease by biomarkers (Figure 2C) clearly showed that most of the miRNAs were known to be involved in colorectal and/or other cancer types. Regulation and proliferation pathway analysis data of miRNAs in tumor vs benign and metastasis vs benign cancer samples, showed most of the miRs were involved in colon cancer and cellular migration (Figures 3A-3E). We further viewed the network analysis of the miRNAs that were involved in signaling pathways. We observed three major pathways were commonly affected in the tumor and metastasis groups (Figures 3F-3H) and only specific to the metastasis group (Figures 3I-3K).
Comparative enrichment analysis of microRNA for Pathway Maps, Gene Ontology, Disease by Biomarker, and Network Processes. MetaCore pathway analysis of small RNA-sequencing data from the tumor and metastatic cancer samples were uploaded to MetaCore servers and most significantly affected pathways analyzed. A: Pathway Maps: Canonical pathway maps were generated, where three out of the top four pathways are involved in colorectal cancer pathogenesis (regulation of miRNAs, role of miRNAs in cell proliferation; and role of miRNAs in migration, survival and angiogenesis). B: Geno Ontology: GO processes were analyzed illustrating that gene silencing of miRNA and post transcriptional gene silencing most impacted. C: Disease status: Significantly involved miRNAs in disease were listed; most of the miRNAs were involved in adenocarcinomas. D: Comparative enrichment analysis: Differential expression of miRNAs in disease groups.

Top regulated Pathway Maps and Network processes. Small RNA-seq data from the tumor and metastasis cancer samples were entered into MetaCore and top regulated pathways and network processes were presented. A to E: Regulation of miRNAs in Colorectal Cancer: Top five highest scored pathway maps linked to colorectal cancers with involved in migration and angiogenesis. Upward thermometers have red color and indicate up‑regulated signals and downward (blue) ones indicate downregulated expression levels of the genes. F-H: Generation of biological networks using comparison Analyze Networks (cAN) algorithm common in both Colorectal Cancer groups: Comparison of the disease group enrichment analysis revealed three commonly affected pathways in both groups. I-K: Generation of biological networks using Analyze Networks (AN) algorithm in metastasis Colorectal Cancer group only: This is a variant of the shortest paths algorithm with main parameters of enrichment with enriched miRNAs prioritized based on the number of fragments of canonical pathways in the networks. Up‑regulated genes are marked with red circles; downregulated with blue circles. The 'checkerboard' color indicates mixed expression of the gene between files or between multiple tags for the same gene.
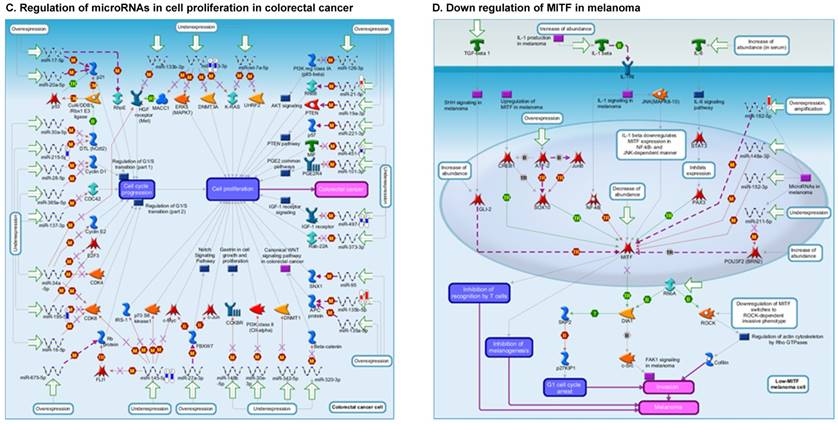
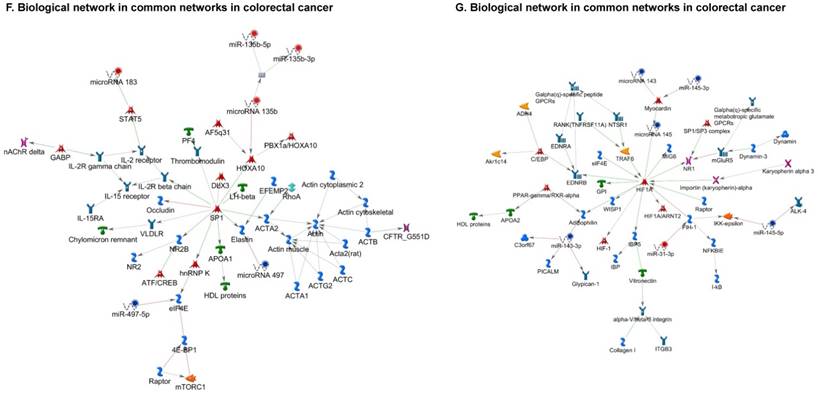
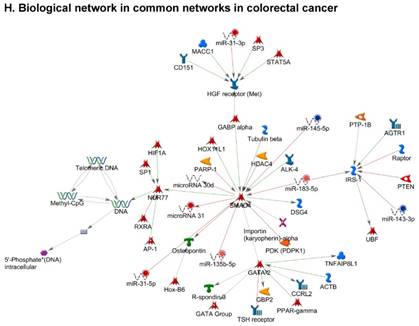
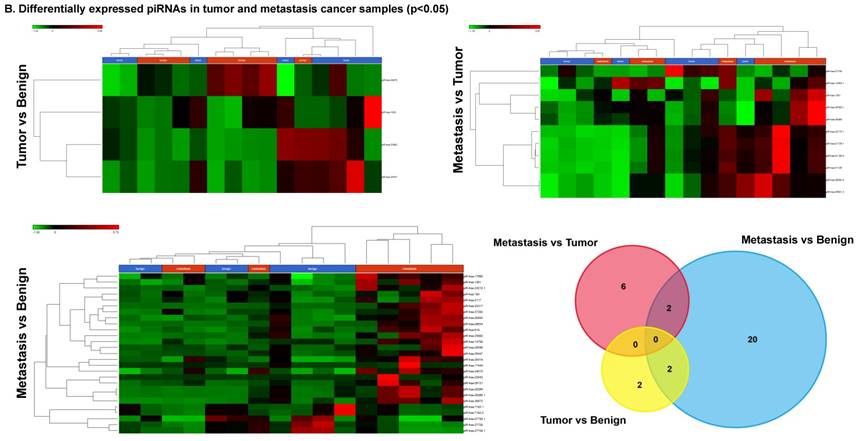
Differentially expressed piRNAs in colorectal cancer tissue samples
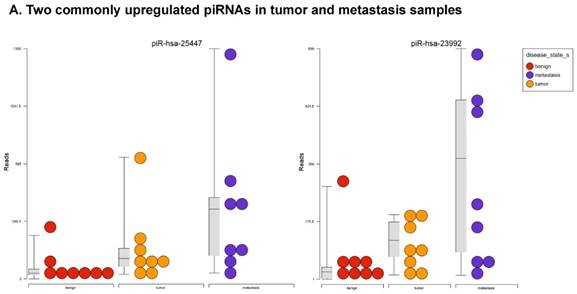
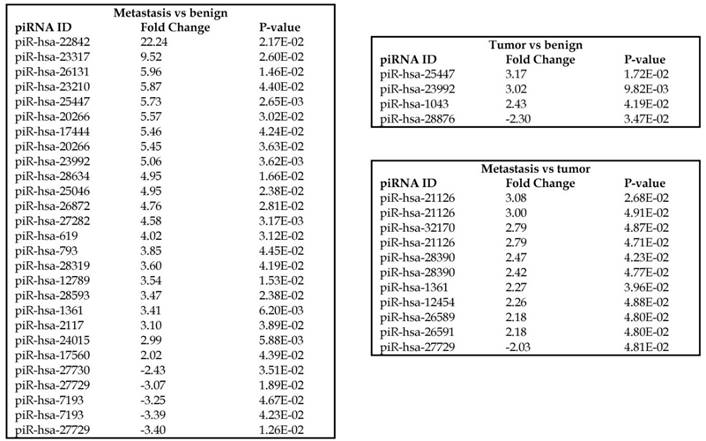
Piwi-interacting RNAs are the largest class of non-coding RNAs [23] recently shown to play important roles in cancer progression. They also represent a third class of small RNA silencers (other two classes are miRNAs and siRNAs) that form RNA-protein complexes by interacting with piwi-proteins. These are required for both epigenetic and post-transcriptional gene silencing of retrotransposons and other genetic elements in germ line cells, particularly during spermatogenesis [24]. We remapped aligned small RNA sequenced data with piRBase annotation and identified six piRNAs (including two piRNAs with alternative transcripts) whose expression levels were statistically significant (p<0.05; Figure 4A, and group wise hierarchical clustering with Venn diagram in Figure 4B). Two piRNAs were commonly upregulated in both tumor and metastasis groups. Tumor vs benign group enriched only four piRNAs which piR-hsa-25447 (3-fold), piR-hsa-23992 (3-fold), and piR-hsa-1043 (2.4-fold) were upregulated and the remaining one downregulated [piR-hsa-28876 (2.3-fold)]. In the metastasis vs benign group, 27 piRNAs were enriched (with multiple transcripts) of which 22 were upregulated [Top five: piR-hsa-22842 (22-fold), piR-hsa-23317 (9.5-fold), piR-hsa-26131 (6-fold) piR-hsa-23210.1 (6-fold) and piR-hsa-25447 (5.7-fold) and the remaining five downregulated [piR-hsa-27729 (3.4-fold), piR-hsa-7193.1 (3.4-fold), piR-hsa-7193.2 (3.2-fold) piR-hsa-27729.1 (3-fold) and piR-hsa-27730.1 (2.4-fold)] (All piRNAs were listed in the Table 2). Recently, more attention is being focused on piRNAs in relation to biologic functions and epigenetic involvement of the biomarker for diagnosis, prognosis, and therapeutic use, which still remains largely unknown. Although piRNA detection in cancer correlates with poorer clinical outcomes suggesting a functional role [25], data available so far is not sufficient to entirely discriminate between a 'passenger' role for the ectopic expression versus a 'driver' role in the pathogenesis of these diseases [25]. Cancer associated piRNAs are especially upregulated in breast cancer (piR-4987, piR-20365, piR-20485, piR-20582 and piR-932) [26, 27], and gastric cancer (piR-823 and piR651) [28, 29]. In kidney cancer both upregulated piRNAs (piR-32051, piR-39894, piR-43607) and down-regulated piRNAs (piR-38756, piR-57125, piR-30924) [30] have been identified. piRNA down-regulation has also been observed in pancreatic cancer (piR-017061) [31], multiple myeloma (piR-823), and lung cancer (piR-L-163) [32].
Differential expression of piRNAs in colorectal cancer: piRNA analysis revealed a total of two piRNAs statistically regulated commonly between two groups. They are (Figure 4A) listed as dot plots. A dendrogram view of all statistically significant piRNAs (p≤0.05) are shown as hierarchical clustering and along with a Venn diagram (Figure 4B).
Differential expression of piRNAs in three groups.
Differentially expressed long non-coding RNAs in colorectal cancer tissue samples
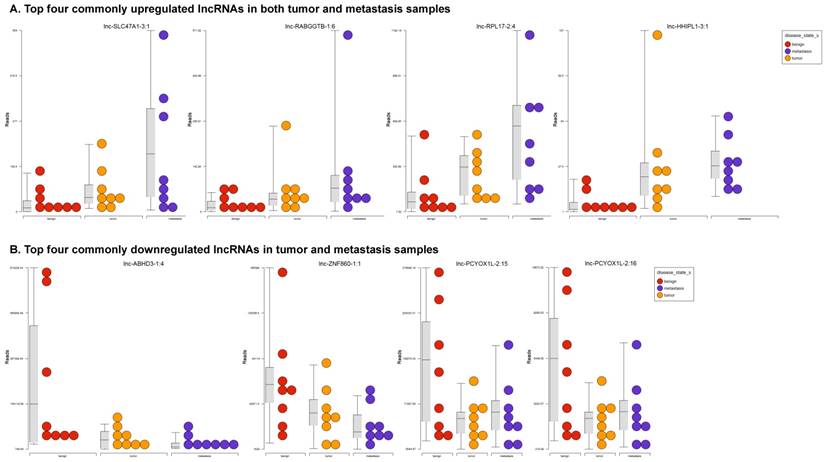
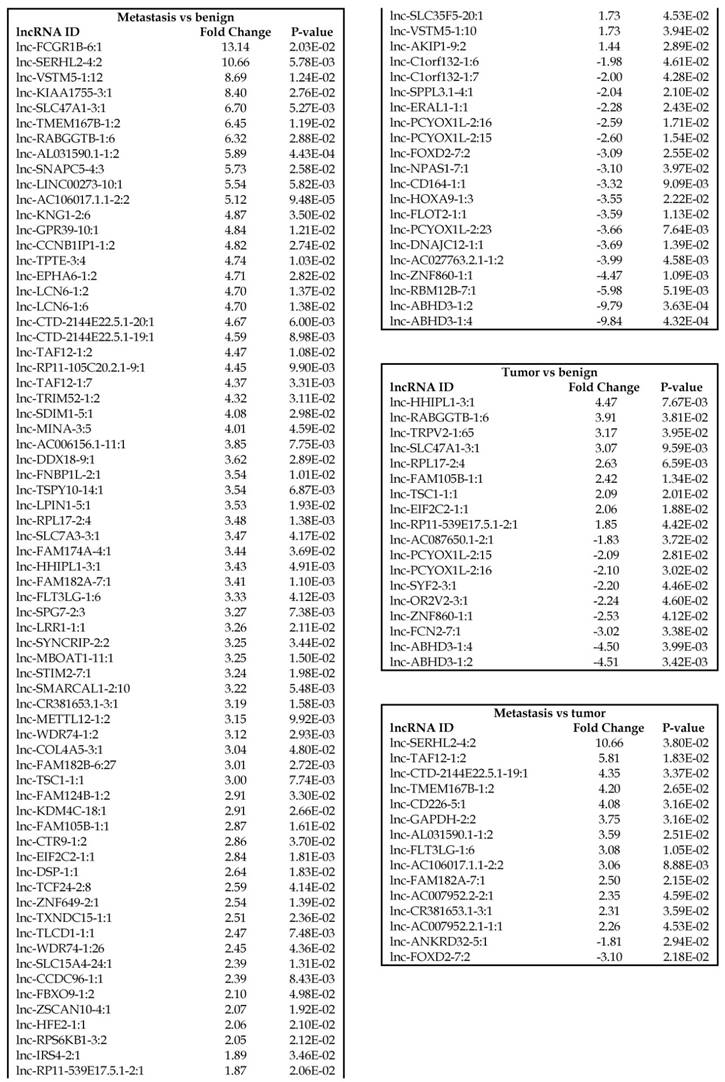
Long non-coding RNAs, larger than 200 nucleotides [24], are the most recent and least characterized ncRNAs. lncRNAs have tissue-specific expression and are expressed in a regulated manner correlating with distinct gene sets that influence cell function [33]. They are also expressed as both tumor suppressors and promoters [34-36]. Our small RNA sequencing data was remapped to identify differentially expressed lncRNAs in colorectal cancer samples. It revealed 18 lncRNAs in tumor vs benign, 89 in metastasis vs benign, and 15 in metastasis vs tumor groups being significantly expressed (p<0.05). The top upregulated ones being lnc-SLC47A1-3:1, lnc-RABGGTB-1:6, lnc-RPL17-2:4 and lnc-HHIPL1-3:1 (Figure 5A); and the top downregulated lncRNAs being lnc-ABHD3-1:4, lnc-ABHD3-1:2, lnc-ZNF860-1:1 and lnc-PCYOX1L-2:15 (Figure 5B). Stage wise hierarchical clustering with Venn diagram for both tumor and metastasis groups is illustrated in Figure 5C along with differential expressed lncRNAs listed in the Table 3. lncRNAs are a heterogeneous group of transcripts, with diverse mechanisms and differential expression in many diseases including tumors [35]. ABHD3 is an alpha/beta hydrolase protein found in a wide range of enzyme domains whose function remains unknown, and multiple transcripts of this gene were downregulated in tumor and metastasis samples in this study. Further exploration is needed to assess their function in cancer and other diseases.
Differential expression of long non-coding RNAs (lncRNAs) in colorectal cancer: Top four upregulated lncRNA were shown in Figure 5A and top four downregulated lncRNAs shown in Figure 5B. Dendrogram view of lncRNAs that are statistically significant (p≤0.05) as a hierarchical tree with stage wise comparison along with a Venn diagram (Figure 5C).
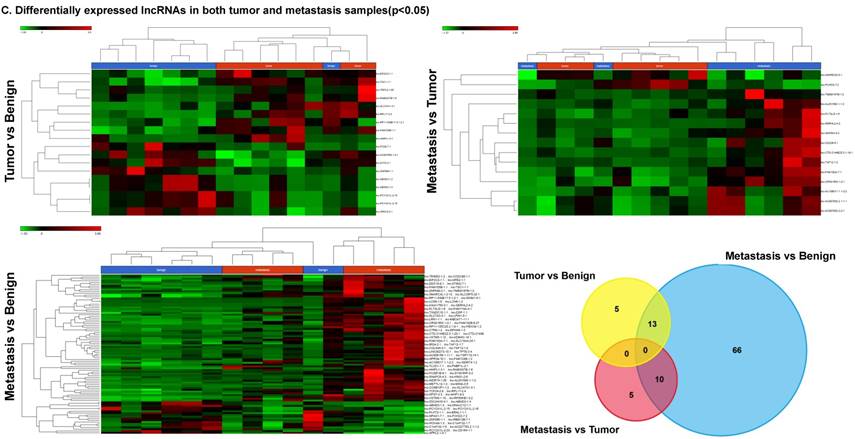
Differential expression of lncRNAs in three groups.
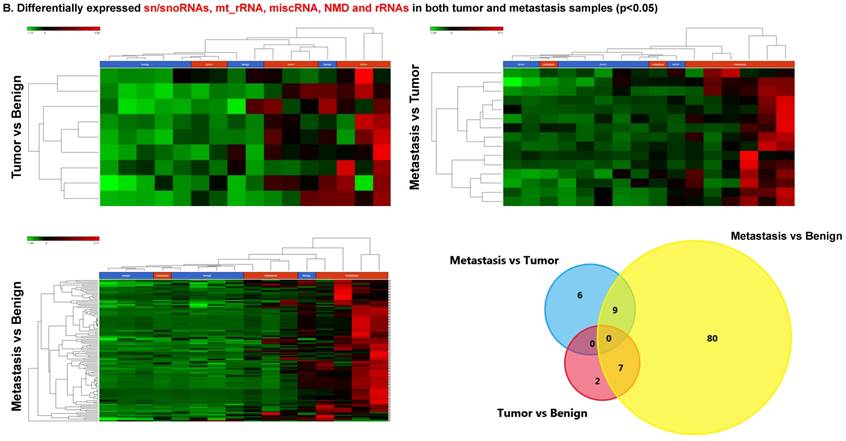
Differentially expressed sn/snoRNAs, mt_rRNA, miscRNA, nonsense mediated decay and rRNAs in colorectal cancer tissue samples
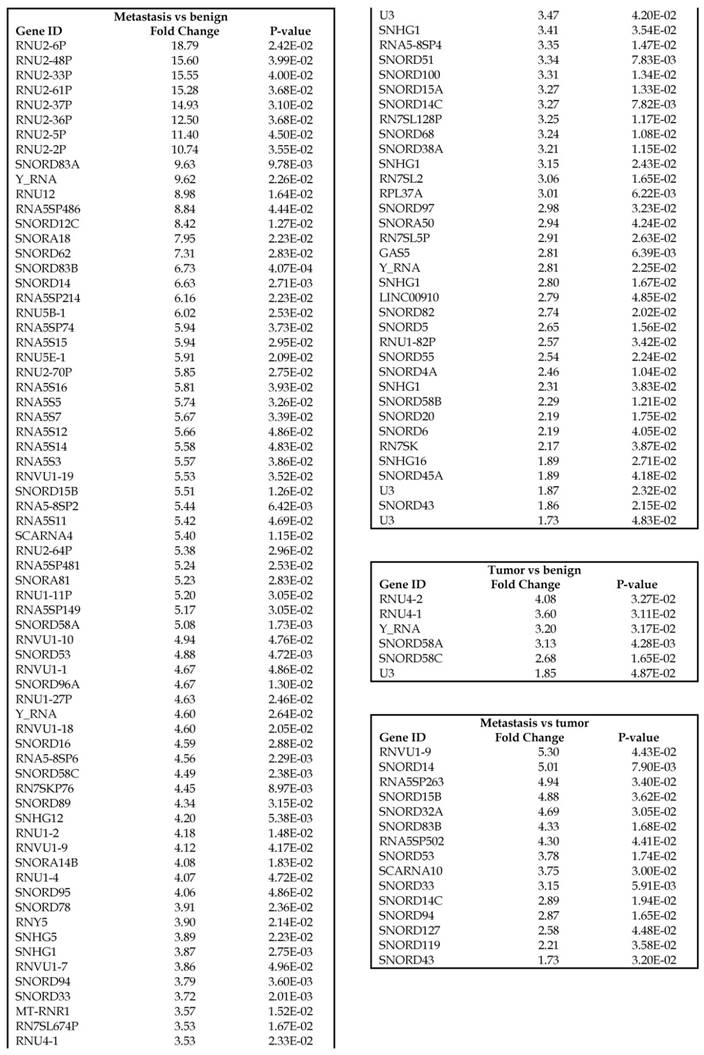
Small nuclear RNAs, form a class of RNA molecules that localize within the nucleus of eukaryotic cells [24]. Their primary function is pre-mRNA processing, for which they are always associated with a set of specific proteins and the complexes are referred to as small nuclear ribonucleoproteins (snRNPs). A subclass of snRNAs called small nucleolar RNAs that localize to the nucleolus are implicated in the maturation of ribosomal and transfer RNA molecules by specific chemical modifications [24]. We remapped the aligned reads to the GenCode database, which contains most of the curated small RNAs. We specifically looked at sn/snoRNAs, mt_rRNA, misc_RNA, nonsense mediated decay (NMD) and rRNAs. All the mentioned sncRNAs were significantly upregulated during CRC disease progression (Figure 6A, and stage wise hierarchical clustering with Venn diagram in Figure 6C). In our investigation, nine differential expression genes were noted in the tumor vs benign group, 104 in metastasis vs benign (with multiple transcripts), and 15 in metastasis vs tumor group. This sncRNA cohort included all sn/snoRNAs, mt_rRNA, misc_RNA, NMD and rRNAs, with most belonging to sn/snoRNAs (listed in the Table 4). Increased expression was noted during metastasis, suggesting potential biomarker use in late stage cancers. Previous studies have shown the RNU family of snRNAs to be expressed in pancreatic, colorectal and lung cancers [37] and decreasing after tumor removal by surgery. Recent studies have also shown that the SNHG (small nucleolar RNA host genes) family of snoRNAs plays a crucial role in cancer. For example, Zhang et al., showed upregulation of SNHG1 in hepatocellular carcinoma, affecting tumor suppressor genes such as p53 and others [38], while Zhao et al., showed downregulation of SNHG5 in gastric cancer [39]. Conversely, in our study, SNHG12 (two transcripts, 4.2-fold) and SNHG1 (3.4-fold) increased. Other snoRNAs, i.e., snoRD14 (5-fold), snoRD15B (5-fold), snoRD32A (4.7-fold) were only upregulated in the metastasis stage (Table 4). Further validation of identified snRNAs are needed to confirm their biological functions and possible use as biomarkers.
Differential expression of sn/snoRNAs, mt_rRNA, miscRNA, nonsense mediated decay and rRNAs in colorectal cancer tissue: small RNA-seq data was analyzed for sn/snoRNAs, mt_rRNA, miscRNA, nonsense mediated decay and rRNAs with GenCode annotation and found nine genes in tumor vs benign, 104 in metastasis vs benign and 15 in metastasis vs tumor groups. Two commonly upregulated sn/snoRNAs are shown in Figure 6A and hierarchical clustering is shown in Figure 6B with an associated Venn diagram.
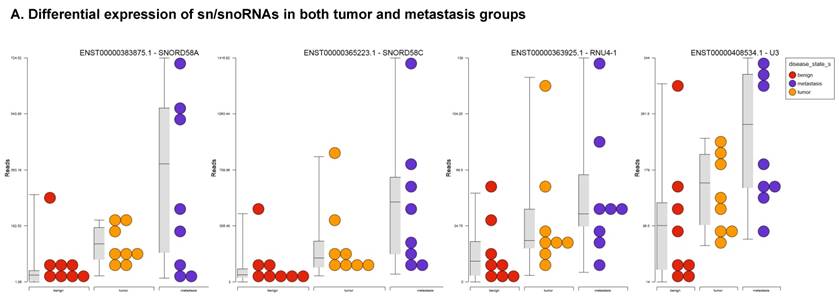
Differential expression of sn/snoRNAs in three groups.
Conclusions
High-throughput sequencing technologies have effectively delineated the crucial roles of non-coding RNAs in normal cellular biology and cancer progression. We have identified several small non-coding RNAs which may be involved in the stepwise progression of normal colon tissue to cancer and subsequent metastatic disease. However, their functions are still relatively unknown. Our analysis can provide a comprehensive list of the miRNAs, piRNAs, lncRNAs and sn/snoRNAs specific to each stage with the commonly expressed genes listed in Tables 1-4. Further understanding and validating will allow us to link these distinct small RNAs with the phenotypic traits seen in the evolution of colorectal cancer, thereby offering a potential for biomarker development. Likewise, further understanding of their roles in cancer signaling pathways may represent a novel target for treatment. Detailed analysis of the individual transcriptome can personalize both prognosis and intervention strategies in colorectal cancer.
Abbreviations
ncRNA: non-coding RNA
sncRNA: small non-coding RNA
miRNA: micro RNA
piRNA: piwi-interacting RNA
lncRNA: long non-coding RNA
snRNA: small nuclear RNA
snoRNA: small nucleolar RNA
mt_rRNA: mitochondrial rRNA
SNHG: small nucleolar RNA host genes
CRC: colorectal cancer
SRA: sequence raw archive
FDR: false discovery rate
NGS: Next Generation Sequencing
GO: Gene ontology
Acknowledgements
We would like to acknowledge Gene Arrays (An entity of Vedic Research, Inc., USA) for MetaCore data analysis free of charge and the Department of Surgery at Penn State University for startup support.
Funding
Funding was provided by the Penn State Department of Surgery.
Ethics
Data presented in the study was download from the NIH data sets (GSE46622) and original authors obtained the formal consents. We did not recruit any human subjects for this study (Not applicable).
Authors contributions
SVK: designed and supervised the study, experimentation, data analysis and manuscript preparation. DJR and AKT: data analysis and manuscript editing. SWH and MM: manuscript editing. All authors have read and approved the final manuscript.
Data availability and materials
Data available at NIH GEO website: project no: PRJNA201245; GEO: GSE46622.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: A Cancer Journal for Clinicians. 2016;66:7-30
2. Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2:919-29
3. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843-54
4. Sai Lakshmi S, Agrawal S. piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Research. 2008;36:D173-D7
5. Scott MS, Ono M, Yamada K, Endo A, Barton GJ, Lamond AI. Human box C/D snoRNA processing conservation across multiple cell types. Nucleic Acids Research. 2011;40:3676-88
6. Cao J. The functional role of long non-coding RNAs and epigenetics. Biological Procedures Online. 2014;16:11
7. Moyano M, Stefani G. piRNA involvement in genome stability and human cancer. Journal of Hematology & Oncology. 2015:8
8. Thomson T, Lin H. The Biogenesis and Function PIWI Proteins and piRNAs: Progress and Prospect. Annual review of cell and developmental biology. 2009;25:355-76
9. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H. et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775-89
10. Scott MS, Ono M. From snoRNA to miRNA: Dual function regulatory non-coding RNAs. Biochimie. 2011;93:1987-92
11. Röhr C, Kerick M, Fischer A, Kühn A, Kashofer K, Timmermann B. et al. High-Throughput miRNA and mRNA Sequencing of Paired Colorectal Normal, Tumor and Metastasis Tissues and Bioinformatic Modeling of miRNA-1 Therapeutic Applications. PLoS ONE. 2013;8:e67461
12. Firmino N, Martinez VD, Rowbotham DA, Enfield KS, Bennewith KL, Lam WL. HPV status is associated with altered PIWI-interacting RNA expression pattern in head and neck cancer. Oral Oncol. 2016;55:43-8
13. Koduru S, Tiwari A, Leberfinger A, Hazard S, Kawasawa Y, Mahajan M. et al. A Comprehensive NGS Data Analysis of Differentially Regulated miRNAs, piRNAs, lncRNAs and sn/snoRNAs in Triple Negative Breast Cancer. Journal of Cancer. 2017;8:578-96
14. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Research. 2014;42:D68-D73
15. Zhang P, Si X, Skogerbø G, Wang J, Cui D, Li Y. et al. piRBase: a web resource assisting piRNA functional study. Database. 2014;2014:bau110
16. Volders P-J, Verheggen K, Menschaert G, Vandepoele K, Martens L, Vandesompele J. et al. An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic Acids Research. 2015;43:D174-D80
17. Sehgal K, Guo X, Koduru S, Shah A, Lin A, Yan X. et al. Plasmacytoid dendritic cells, interferon signaling, and FcgammaR contribute to pathogenesis and therapeutic response in childhood immune thrombocytopenia. Sci Transl Med. 2013;5:193ra89
18. Taniguchi K, Sakai M, Sugito N, Kumazaki M, Shinohara H, Yamada N. et al. PTBP1-associated microRNA-1 and -133b suppress the Warburg effect in colorectal tumors. Oncotarget. 2016;7:18940-52
19. Wu W, Wang Z, Yang P, Yang J, Liang J, Chen Y. et al. MicroRNA-135b regulates metastasis suppressor 1 expression and promotes migration and invasion in colorectal cancer. Molecular and Cellular Biochemistry. 2014;388:249-59
20. Nagel R, le Sage C, Diosdado B, van der Waal M, Oude Vrielink JAF, Bolijn A. et al. Regulation of the Adenomatous Polyposis Coli Gene by the miR-135 Family in Colorectal Cancer. Cancer Research. 2008;68:5795-802
21. Bandrés E, Cubedo E, Agirre X, Malumbres R, Zárate R, Ramirez N. et al. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol Cancer. 2006;5:29
22. O'Day E, Lal A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010;12:201
23. Fu A, Jacobs DI, Hoffman AE, Zheng T, Zhu Y. PIWI-interacting RNA 021285 is involved in breast tumorigenesis possibly by remodeling the cancer epigenome. Carcinogenesis. 2015;36:1094-102
24. Morceau F, Chateauvieux S, Gaigneaux A, Dicato M, Diederich M. Long and Short Non-Coding RNAs as Regulators of Hematopoietic Differentiation. International Journal of Molecular Sciences. 2013;14:14744-70
25. Moyano M, Stefani G. piRNA involvement in genome stability and human cancer. Journal of Hematology & Oncology. 2015;8:38
26. Huang G, Hu H, Xue X, Shen S, Gao E, Guo G. et al. Altered expression of piRNAs and their relation with clinicopathologic features of breast cancer. Clinical and Translational Oncology. 2013;15:563-8
27. Zhang H, Ren Y, Xu H, Pang D, Duan C, Liu C. The expression of stem cell protein Piwil2 and piR-932 in breast cancer. Surgical Oncology. 2013;22:217-23
28. Cheng J, Deng H, Xiao B, Zhou H, Zhou F, Shen Z. et al. piR-823, a novel non-coding small RNA, demonstrates in vitro and in vivo tumor suppressive activity in human gastric cancer cells. Cancer Letters. 2012;315:12-7
29. Cui L, Lou Y, Zhang X, Zhou H, Deng H, Song H. et al. Detection of circulating tumor cells in peripheral blood from patients with gastric cancer using piRNAs as markers. Clinical Biochemistry. 2011;44:1050-7
30. Busch J, Ralla B, Jung M, Wotschofsky Z, Trujillo-Arribas E, Schwabe P. et al. Piwi-interacting RNAs as novel prognostic markers in clear cell renal cell carcinomas. Journal of Experimental & Clinical Cancer Research: CR. 2015;34:61
31. Müller S, Raulefs S, Bruns P, Afonso-Grunz F, Plötner A, Thermann R. et al. Next-generation sequencing reveals novel differentially regulated mRNAs, lncRNAs, miRNAs, sdRNAs and a piRNA in pancreatic cancer. Mol Cancer. 2015;14:94
32. Mei Y, Wang Y, Kumari P, Shetty AC, Clark D, Gable T. et al. A piRNA-like small RNA interacts with and modulates p-ERM proteins in human somatic cells. Nature Communications. 2015;6:7316
33. Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223-7
34. Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D. et al. A large intergenic non-coding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409-19
35. Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21:1253-61
36. Zheng GXY, Do BT, Webster DE, Khavari PA, Chang HY. Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat Struct Mol Biol. 2014;21:585-90
37. Köhler J, Schuler M, Gauler TC, Nöpel-Dünnebacke S, Ahrens M, Hoffmann A-C. et al. Circulating U2 small nuclear RNA fragments as a diagnostic and prognostic biomarker in lung cancer patients. Journal of Cancer Research and Clinical Oncology. 2016;142:795-805
38. Zhang M, Wang W, Li T, Yu X, Zhu Y, Ding F. et al. Long noncoding RNA SNHG1 predicts a poor prognosis and promotes hepatocellular carcinoma tumorigenesis. Biomedicine & Pharmacotherapy. 2016;80:73-9
39. Zhao L, Guo H, Zhou B, Feng J, Li Y, Han T. et al. Long non-coding RNA SNHG5 suppresses gastric cancer progression by trapping MTA2 in the cytosol. Oncogene. 2016;35:5770-80
Author contact
![]() Corresponding author: Srinivas V Koduru, PhD., Division of Plastic Surgery, Department of Surgery, College of Medicine, Pennsylvania State University, 500 University Drive, Hershey, PA 17033 Email: skodurupsu.edu Phone: 717-531-4332
Corresponding author: Srinivas V Koduru, PhD., Division of Plastic Surgery, Department of Surgery, College of Medicine, Pennsylvania State University, 500 University Drive, Hershey, PA 17033 Email: skodurupsu.edu Phone: 717-531-4332