ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2017; 5:77-82. doi:10.7150/jgen.20216 This volume Cite
Research Paper
A glimpse into the genetic basis of symbiosis between Hydrogenophaga and their helper strains in the biodegradation of 4-aminobenzenesulfonate
Kangsan Kim1,2, Han Ming Gan1,2,3 ![]()
1. Genomics Facility, Tropical Medicine and Biology Platform, Monash University Malaysia, Jalan Lagoon Selatan, Bandar Sunway, 47500 Selangor, Malaysia
2. School of Science, Monash University Malaysia, Jalan Lagoon Selatan, Bandar Sunway, 47500 Selangor, Malaysia
3. Centre for Integrative Ecology, School of Life and Environmental Sciences, Deakin University, Pigdons Road, Waurn Ponds, Victoria 3216 Australia
Received 2017-3-23; Accepted 2017-5-29; Published 2017-7-10
Abstract

We report the whole genome sequences of Hydrogenophaga intermedia S1 and Agrobacterium radiobacter S2, the first reported bacterial co-culture capable of degrading 4-aminobenzenesulfonate (4-ABS), a recalcitrant industrial waste product. To gain insights into the genetic basis for the syntrophic interaction between this symbiotic pair and also another recently reported Hydrogenophaga associated co-culture, Hydrogenophaga sp. PBC and Ralstonia sp. PBA, we performed detailed genetic analysis of these four strains focusing on the metabolic pathways associated with biotin, para-aminobenzoic acid (pABA), and protocatechuate metabolism. Both assembled Hydrogenophaga draft genomes are missing a majority of the genetic components associated in the biosynthetic pathway of pABA and biotin. Interestingly, a fused pABA synthase was found in R. sp PBA but not in A. radiobacter S2. Furthermore, using whole genome data, the taxonomic classification of R. sp. PBA and A. radiobacter S2 (both previously inferred from 16S rRNA gene) was re-investigated, providing new evidence to propose for their re-classification at the genus and species level, respectively
Keywords: p-aminobenzoic acid, biotin, co-culture, phylogenomics, Hydrogenophaga, Agrobacterium, 4-aminobenzenesulfonate
Introduction
4-aminobenzenesulfonate (4-ABS) is a type of sulfonated aromatic amines that serve as key intermediates in the manufacturing of dyes, sulfa drugs, brighteners and pesticides. Presence of sulfonate moiety on aromatic ring renders the compound recalcitrant against biodegradation in activated sludge by aromatics-degrading bacteria [1-3]. In addition, the inhibitory effects of 4-ABS on prokaryotic folate metabolism presents another barrier to its utilization by common bacterial colonies [4]. Not surprisingly, microbial degradation of 4-ABS is a rare occurrence among microbiomes found in natural soils, sediments and in activated sludge [5]. The first reported, and likely one of the most efficient microbial biodegradation of 4-ABS was reported in a co-culture consisting of Hydrogenophaga intermedia S1 and Agrobacterium radiobacter S2 isolated from Germany [1]. 15 years later, a co-culture consisting of another Hydrogenophaga strain but with a beta-proteobacterium, Ralstonia sp. PBA, was isolated from textile wastewater in Malaysia similarly demonstrating efficient 4-ABS biodegradation ability [5, 6].
In strains S1/S2, it was demonstrated that the metabolites secreted by strain S2 e.g. p-aminobenzoate (pABA) and biotin, served as growth factors for strain S1, enabling the strain to degrade 4-ABS [6]. The first catabolic step of 4-ABS by S1 involves deamination of the compound to 4-sulfocatechol (4SC) [1]. The downstream reaction of 4-ABS degradation is characterized by ring cleavage of 4SC catalyzed by the combined actions of type II protocatechuate 3,4-dioxygenase alpha and protocatechuate 3,4-dioxygenase beta unit which are encoded by pcaG2 and pcaH2 respectively [7]. The catabolism of 4-SC involves ring cleavage and desulfonation catalyzed by PcaB2 (a modified 3-carboxy-cis,cis-muconate-lactonizing enzyme) and 4-sulfomuconolactone hydrolase, respectively, leading to the generation of maleylacetate that can be channeled into the tricarboxylic acid cycle [7-10].
The symbiotic relationship between Hydrogenophaga sp. PBC and Ralstonia sp. PBA is slightly different in that the helper strain PBA was not able to utilize 4-sulfocatechol as its sole carbon source and may depend on other compounds generated from the later step of 4-ABS metabolism to maintain growth [5]. Genomic resources for Hydrogenophaga sp. PBC and Ralstonia sp. PBA are now available [11, 12] and preliminary genomic analysis has shown that strain PBA contains two copies of pabB gene potentially allowing it to overproduce this growth factor. However, it remains unclear if similar genetic feature is present in A. radiobacter S2.
The species designation of strain S2 as A. radiobacter based on 16S rRNA phylotyping may be questionable given its atypical isolation source e.g. non-plant host. This is further complicated by the current lack of taxonomic consensus on the nomenclature and classification of Agrobacterium species [13]. Similarly, the genus designation of Ralstonia sp. PBA may benefit from additional investigation given its potentially unreliable classification based on 16S rRNA and also its substantially smaller genome size compared to other sequenced strains in the genus Ralstonia [14, 15].
In this study, we aim to (1) sequence the genome of Hydrogenophaga intermedia S1T and Agrobacterium radiobacter S2, the first 4-ABS degrading co-culture strains to be reported, (2) understand the genetic basis for symbiotic relationship among 4-ABS degrading co-cultures, and (3) revisit the taxonomic status of Agrobacterium radiobacter S2 and Ralstonia sp. PBA using recent genomic concepts for taxonomic demarcation.
Materials and Methods
Whole genome sequencing and genome assembly
Purified genomic DNA of strains S1 and S2 were tagmented with Nextera XT (Illumina, San Diego, CA) and sequenced on the MiSeq desktop sequencer located at the Monash University Malaysia Genomics Facility using a 2 × 150 bp paired-end run configuration. Nextera adapter sequences in raw FASTQ sequence reads were trimmed using Trimmomatic software v0.35 [16], error-corrected and assembled de novo into contigs with SPAdes genome assembler v3.5.0 [17]. The assembled contigs were extended and scaffolded using SSPACE v.3.0 [18]; gaps were removed with GapFiller v1.10 [19]. Average nucleotide identity (ANI) was calculated using JSpecies v1.2.1 [20].
Phylogenomic analysis and protein domain identification
Protein coding sequences were predicted from the assembled contigs using Prodigal v1.20, a gene prediction algorithm for prokaryotes [21] and subsequently utilized for phylogenomics tree construction with PhyloPhlAn. The percentage of conserved proteins (POCP) matrix was computed as previously reported [22]. Protein domain identification was performed using HMMsearch v3.1 [23] against selected TIGRFAM and Pfam profiles (Fig. 1A and B). For TIGRFAM profiles [24], significant hits were filtered using both trusted cutoff (the lowest-scoring known true positive that is above all known false positives) and noise cutoff (the highest-scoring known false positive) settings while for Pfam profiles [25], an E-value cutoff of 1e-5 was used [26].
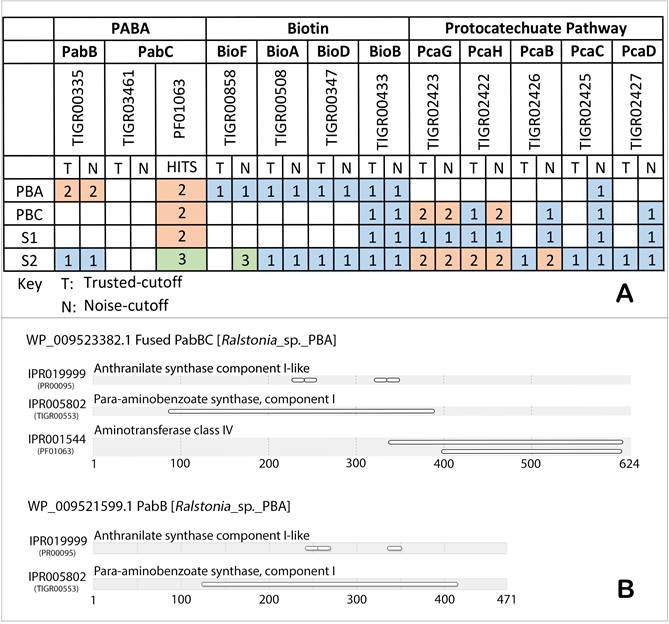
(A) Putative genetic catalogues of biosynthetic and metabolic pathways of pABA, biotin and protocatechuate in strain PBC, PBA, S1 and S2, as searched against TIGRFAM [24] and Pfam [25] databases. Number of hits designated to each of the strains was color annotated accordingly. (B) Fused PabBC and PabB protein domains identification in strain PBA, as searched against InterPro databases via InterProScan [26].
Results and Discussion
Comparative analysis of genes involved in the biosynthesis of p-aminobenzoate
The absence of gene coding aminodeoxychorismate synthase (PabB) critical for the synthesis of pABA [27, 28] in both annotated genomes of strains S1 and PBC corroborates with their requirement for pABA to maintain growth in axenic culture (Figure 1A). Interestingly, two positive TIGRfam protein domain hits were detected from the annotated genes of strain PBA for PabB (TIGR00553) which catalyzes the formation of 4-amino-4-deoxychorismate from ammonia and chorismate [29]. However, the HMM scores for the TIGR03461 hits corresponding to PabC, the catalytic enzyme involved in breakdown of 4-amino-4-deoxychorismate into pABA and pyruvate [30] were below the noise cutoff. Using an alternative HMM profile, PF01063 containing the amino-transferase class IV protein domain typically associated with but not specific to PabC, two significant hits were obtained of which one of them interestingly hit to a PabB protein (WP_009523382.1, contig AKCV01000025), indicating a potentially fused PabBC enzyme (Figure 1B). On the other hand, the second identified PabB (WP_009521599.1, contig AKCV01000019.1) was not genetically linked to any PF01063 domain-containing protein (Figure 1B).
Fused PabBC enzyme has been previously reported in several strains of gram-positive and gram-negative bacteria such as Lactococcus lactis, Streptococcus pyogenes and Helicobacter pylori [31]. Group of proteins that execute functionally reciprocal interactions incline to fuse together, giving rise to a protein modularity with defined domains of biological functions [32, 33]. In addition, physical amalgamation of such kind endows selective advantage over allosteric gene regulation, metabolic features, protein size and many more - conferring a competitive edge over the survival of a strain [34]. In the case of fused PabBC in Ralstonia sp. PBA, this can potentially lead to the overproduction of pABA which can sustain the requirement of a pABA auxotroph partner e.g. Hydrogenophaga sp. PBC leading to the establishment of a stable symbiotic relationship. Further study is necessary however to confirm the function of both TIGR00553 domain-containing proteins and to measure the catalytic efficiency of the fused PabBC in pABA synthesis.
On the contrary, A. radiobacter S2 contains only one TIGRfam hit to PabB while the authentic PabC for this bacterium could not be confidently identified as none of the three genes coding for protein with hit to PF01063 was located on the same contigs as pabB. Surprisingly, subsequent search for TIGR03641 hits within the annotated genes of members from the genus Agrobacterium or even the known pabC-containing Pseudomonas aeruginosa strain PAO1 in Uniprot (as of 14 March 2017) returned zero hit (data not shown), suggesting the need to revise HMM scoring profile and threshold for TIGR03641 and/or the possible existence of alternative aminodeoxychorismate lyase as previously reported in Chlamydia [35]. Although we could not find genomic evidence to suggest enhanced pABA synthesis in strain S2, profligate synthesis of biotin instead has been demonstrated in wild-type A. tumefaciens (identical to A. radiobacter [36]) with biotin production per viable cell that is 20-and 60-fold greater than that of E.coli and B. subtilis, respectively. Such trait was subsequently suggested to be useful to A. tumefaciens in that the recipients of the cofactor will aid the growth and survival of A. tumefaciens [37].
Genes coding for enzymes involved in the biosynthesis of biotin are severely lacking in Hydrogenophaga intermedia S1 and Hydrogenophaga sp. PBC
Interestingly, while it was suggested that strains S1 and PBC were auxotrophs to biotin [5, 6], TIGRFAM search revealed the presence of a biotin synthase (BioB, EC:2.8.1.6) in their annotated genes (Fig 1A). Biotin serves as an integral cofactor in carbon fixation reactions [38], and BioB catalyzes the terminal-stage reaction of biotin synthesis through which it generates biotin from its precursor dethiobiotin [39]. Nonetheless, biotin biosynthesis encompasses myriads of biochemical reactions requiring other associated enzymes such as 8-amino-7-oxononanoate synthase, adenosylmethionine-8-amino-7-oxononanoata aminotransferase and dethiobiotin synthase which were not detected in the annotated genomes of strains S1 and PBC [38].
Protocatechuate and 4-sulfocatechol catabolism among helper strains
Ralstonia sp. PBA lacked a substantial portion of integral enzymes associated with the catabolism of protocatechuate and/or 4-sulfocatechol (Fig. 1A) corroborating with previous biochemical results [5, 40]. Unlike Ralstonia sp. PBA, A radiobacter S2 could grow on 4-sulfocatechol secreted by H. intermedia S1 during 4-ABS catabolism as its sole carbon source [1]. Two types of protocatechuate dioxygenases were identified in the annotated genome of A. radiobacter S2 (Fig. 1B) which is consistent with previous molecular work showing that both variants could degrade protocatechuate with high efficiency but only one of them (protocatechuate 3,4-dioxygenase type II), showed activity towards 4-sulfocatechol [41].
Taxonomic revision of Agrobacterium radiobacter S2 and Ralstonia sp. PBA
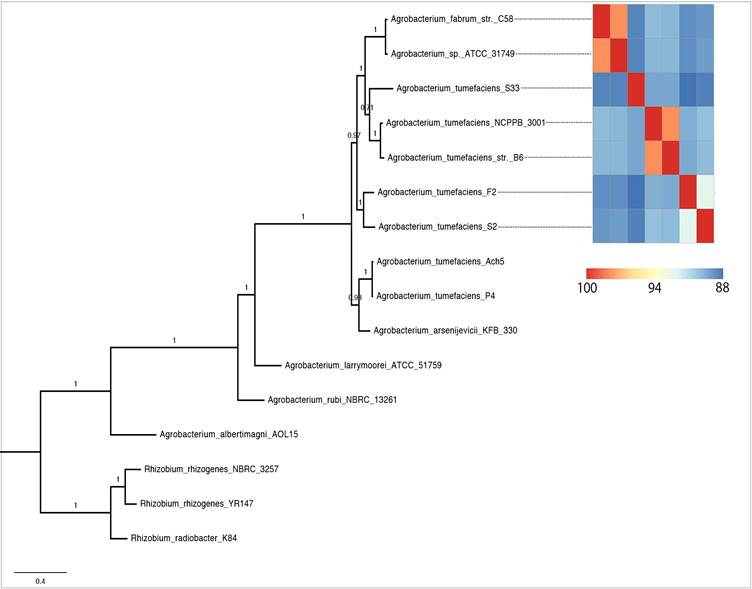
Phylogenomic analysis (Fig. 2) showed that strains currently assigned as A. tumefaciens (identical to A. radiobacter [36]) shared a common ancestor with A. fabrum and A. sp. ATCC31974. Among them, A. tumefaciens F2 was sister taxa to strain S2 with maximal SH-like bootstrap support. Surprisingly, with a suggested ANI parameter of ~95-96% for species delineation [20], 5 distinct genospecies could be identified for members within this clade. Two genomic clusters represented by two genome pairs e.g. (i) A. tumefaciens NCPPB3001-A. tumefaciens B6 and (ii) A. fabrum C58-A. sp. ATCC31749 could be designated with a valid Agrobacterium species name [20]. For strain S2 however, given the lack of >95% ANIm similarity to known Agrobacterium species, its current taxonomic assignment as A. radiobacter is not valid and we propose a more conservative classification of strain S2 as Agrobacterium sp. pending future taxonomic description.
Maximum likelihood tree of the genus Agrobacterium rooted with members of the genus Rhizobium as the outgroup. Heatplot next to the tree is ANIm matrix generated from the 7 selected Agrobacterium strains.
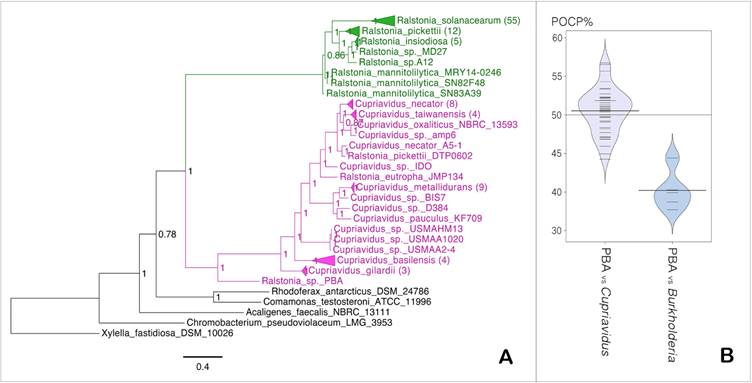
The basal position of Ralstonia sp. PBA within the Cupriavidus clade indicates that it is more closely related to the genus Cupriavidus than Ralstonia (Fig. 3A). Pairwise comparison of the whole proteome of strain PBA against members of genus Cupriavidus generated median POCP value of 51% (Fig. 1D), which slightly surpassed the genus demarcation threshold of 50% proposed by Qin et al., 2014 [22] (Fig. 3B). Taken together, the data seemed to consolidate the proposed phylogenetic affinity of strain PBA to the genus Cupriavidus instead of Ralstonia but considering the intertwined biography of the genus Cupriavidus with several other β-proteobacteria (Ralstonia, Wautersia, Alcaligenes), genus classification for this group of bacteria could be fairly misleading [42]. In fact, strains that retain several basonyms, such as Ralstonia eutropha - previously known as Cupriavidus metallidurans or Alcaligenes eutrophus, well represent the history of intermingled nomenclature and taxonomy of the genus Ralstonia [42]. With the availability of more genomic resources, a more comprehensive genomic analysis of members of these related genera will be beneficial which is beyond the scope of this study.
(A) Maximum likelihood tree of the genera Cupriavidus and Ralstonia. Values in nodes indicate SH-like support values, numbers in brackets indicate number of branches collapsed and scale bar indicates number of amino acid substitutions per site (B) Bean plot of POCP data calculated between PBA vs members of the genus Cupriavidus and Burkholderia.
Data Availability
The assembled draft genomes of strains S1 and S2 are 5 MB (124 contigs) and 5.7 MB (60 contigs), respectively, and have been deposited into the NCBI database under the accession numbers CCAE01000000 and CCAN01000000, respectively. Whole genome sequencing for strains PBA and PBC has been previously reported [11, 12].
Acknowledgements
We thank the Monash University Malaysia Tropical and Medicine Biology Platform for financial and infrastructure support. We also are grateful to Dr Andreas Stolz for the provision of genomic DNA for Hydrogenophaga intermedia S1 and Agrobacterium radiobacter S2.
Competing Interests
The authors have declared that no competing interests exist.
References
1. Feigel BJ, Knackmuss HJ. Syntrophic interactions during degradation of 4-aminobenzenesulfonic acid by a two species bacterial culture. Arch Microbiol. 1993;159:124-30
2. Rozgaj RG. Total degradation of 6-aminonaphthalene-2-sulfonic acid by a mixed culture consisting of different bacterial genera. FEMS Microbiol Ecol. 1992;86:229-36
3. Alexander M, Lustigham B. Effect of chemical structure on microbial degradation of substituted benzenes. Journal of Agricultural Food Chemistry. 1996;14:410-3
4. Brown GM. Inhibition by sulfonamides of the biosynthesis of folic acid. Int J Lepr Other Mycobact Dis. 1967;35:580-9
5. Gan HM, Shahir S, Ibrahim Z, Yahya A. Biodegradation of 4-aminobenzenesulfonate by Ralstonia sp. PBA and Hydrogenophaga sp. PBC isolated from textile wastewater treatment plant. Chemosphere. 2011;82:507-13
6. Dangmann E, Stolz A, Kuhm AE, Hammer A, Feigel B, Noisommit-Rizzi N. et al. Degradation of 4-aminobenzenesulfonate by a two-species bacterial coculture. Physiological interactions between Hydrogenophaga palleronii S1 and Agrobacterium radiobacter S2. Biodegradation. 1996;7:223-9
7. Buchan A, Neidle EL, Moran MA. Diverse organization of genes of the beta-ketoadipate pathway in members of the marine Roseobacter lineage. Appl Environ Microbiol. 2004;70:1658-68
8. Halak S, Basta T, Bürger S, Contzen M, Wray V, Pieper DH. et al. 4-Sulfomuconolactone Hydrolases from Hydrogenophaga intermedia S1 and Agrobacterium radiobacter S2. Journal of Bacteriology. 2007;189:6998-7006
9. Contzen M, Burger S, Stolz A. Cloning of the genes for a 4-sulphocatechol-oxidizing protocatechuate 3,4-dioxygenase from Hydrogenophaga intermedia S1 and identification of the amino acid residues responsible for the ability to convert 4-sulphocatechol. Molecular Microbiology. 2001;41:199-205
10. Halak S, Basta T, Burger S, Contzen M, Stolz A. Characterization of the genes encoding the 3-carboxy-cis,cis-muconate-lactonizing enzymes from the 4-sulfocatechol degradative pathways of Hydrogenophaga intermedia S1 and Agrobacterium radiobacter S2. Microbiology. 2006;152:3207-16
11. Gan HM, Chew TH, Tay YL, Lye SF, Yahya A. Genome sequence of Hydrogenophaga sp. strain PBC, a 4-aminobenzenesulfonate-degrading bacterium. J Bacteriol. 2012;194:4759-60
12. Gan HM, Chew TH, Tay YL, Lye SF, Yahya A. Genome sequence of Ralstonia sp. strain PBA, a bacterium involved in the biodegradation of 4-aminobenzenesulfonate. J Bacteriol. 2012;194:5139-40
13. Young JM, Kuykendall LD, Martinez-Romero E, Kerr A, Sawada H. Classification and nomenclature of Agrobacterium and Rhizobium. Int J Syst Evol Microbiol. 2003;53:1689-95
14. Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol. 2007;45:2761-4
15. Fox GE, Wisotzkey JD, Jurtshuk P Jr. How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Bacteriol. 1992;42:166-70
16. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20
17. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455-77
18. Boetzer M HC, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578-9
19. Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13:R56
20. Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106:19126-31
21. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119
22. Qin QL, Xie BB, Zhang XY, Chen XL, Zhou BC, Zhou J. et al. A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol. 2014;196:2210-5
23. Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755-63
24. Haft DH, Selengut JD, Richter RA, Harkins D, Basu MK, Beck E. TIGRFAMs and Genome Properties in 2013. Nucleic Acids Res. 2013;41:D387-95
25. Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279-85
26. Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236-40
27. Viswanathan VK, Green JM, Nichols BP. Kinetic characterization of 4-amino 4-deoxychorismate synthase from Escherichia coli. J Bacteriol. 1995;177:5918-23
28. Ye QZ, Liu J, Walsh CT. p-Aminobenzoate synthesis in Escherichia coli: purification and characterization of PabB as aminodeoxychorismate synthase and enzyme X as aminodeoxychorismate lyase. Proc Natl Acad Sci U S A. 1990;87:9391-5
29. Rusnak F, Liu J, Quinn N, Berchtold GA, Walsh CT. Subcloning of the enterobactin biosynthetic gene entB: expression, purification, characterization and substrate specificity of isochorismatase. Biochemistry. 1990;29:1425-35
30. Nichols BP, Seibold AM, Doktor SZ. para-aminobenzoate synthesis from chorismate occurs in two steps. The Journal of Biological Chemistry. 1989;264:8597-601
31. Wegkamp A, van Oorschot W, de Vos WM, Smid EJ. Characterization of the role of para-aminobenzoic acid biosynthesis in folate production by Lactococcus lactis. Appl Environ Microbiol. 2007;73:2673-81
32. Edward M. Marcotte MP, Ho-Leung Ng, Danny W. Rice, Todd O. Yeates, David Eisenberg. Detecting Protein Function and Protein-Protein Interactions from Genome Sequences. Science. 1999:5428
33. Wang ML, Caetano-Anollés G. The Evolutionary Mechanics of Domain Organization in Proteomes and the Rise of Modularity in the Protein World. Structure. 2009;17:66-78
34. Hashimoto K, Nishi H, Bryant S, Panchenko AR. Caught in self-interaction: evolutionary and functional mechanisms of protein homooligomerization. Phys Biol. 2011;8:035007
35. Adams NE, Thiaville JJ, Proestos J, Juárez-Vázquez AL, McCoy AJ, Barona-Gómez F. et al. Promiscuous and adaptable enzymes fill “holes” in the tetrahydrofolate pathway in Chlamydia species. mBio. 2014;5:e01378-14
36. Sawada H, Ieki H, Oyaizu H, Matsumoto S. Proposal for rejection of Agrobacterium tumefaciens and revised descriptions for the genus Agrobacterium and for Agrobacterium radiobacter and Agrobacterium rhizogenes. Int J Syst Bacteriol. 1993;43:694-702
37. Feng Y, Zhang H, Cronan JE. Profligate biotin synthesis in alpha-proteobacteria - a developing or degenerating regulatory system? Mol Microbiol. 2013;88:77-92
38. Knowles JR. The mechanism of biotin-dependent enzymes. Annu Rev Biochem. 1989;58:195-221
39. Zhang S, Sanyal I, Bulboaca GH, Rich A, Flint DH. The gene for biotin synthase from Saccharomyces cerevisiae: cloning, sequencing, and complementation of Escherichia coli strains lacking biotin synthase. Arch Biochem Biophys. 1994;309:29-35
40. Gan HM, Ibrahim Z, Shahir S, Yahya A. Identification of genes involved in the 4-aminobenzenesulfonate degradation pathway of Hydrogenophaga sp. PBC via transposon mutagenesis. FEMS Microbiol Lett. 2011;318:108-14
41. Contzen M, Stolz A. Characterization of the Genes for Two Protocatechuate 3,4-Dioxygenases from the 4-Sulfocatechol-Degrading Bacterium Agrobacterium radiobacter Strain S2. Journal of Bacteriology. 2000;182:6123-9
42. Vandamme P, Coenye T. Taxonomy of the genus Cupriavidus: a tale of lost and found. Int J Syst Evol Microbiol. 2004;54:2285-9
Author contact
![]() Corresponding author: Han Ming Gan, Email: han.ganedu.au, Address: Room 4.338, Level 4, Building KA, School of Life and Environmental Sciences, Deakin University, Pigdons Road, Waurn Ponds, Victoria 3216 Australia
Corresponding author: Han Ming Gan, Email: han.ganedu.au, Address: Room 4.338, Level 4, Building KA, School of Life and Environmental Sciences, Deakin University, Pigdons Road, Waurn Ponds, Victoria 3216 Australia