ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2024; 12:58-70. doi:10.7150/jgen.93243 This volume Cite
Research Paper
The impact of Elaeagnus angustifolia root exudates on Parafrankia soli NRRL B-16219 exoproteome
Ikram kammoun1, Guylaine Miotello2, Karim Ben Slama3, Jean Armengaud2, Faten Ghodhbane-Gtari1,4, Maher Gtari1 ![]()
1. Department of Biological and Chemical Engineering USCR Molecular Bacteriology and & Genomics, National Institute of Applied Sciences and Technology, University of Carthage, Tunis, Tunisia.
2. Département Médicaments et Technologies pour la Santé (DMTS), CEA, INRAE, Université Paris-Saclay, SPI, 30200 Bagnols sur Cèze, France.
3. Higher Institute of Applied Biological Sciences, Laboratory of Bioresources, Environment, and Biotechnology, University of Tunis El Manar, Tunis, Tunisia.
4. Higher Institute of Biotechnology of Sidi Thabet, University of La Manouba, Sidi Thabet, Tunisia.
Received 2024-1-1; Accepted 2024-4-21; Published 2024-5-11
Abstract

Root exudates from host plant species are known to play a critical role in the establishment and maintenance of symbiotic relationships with soil bacteria. In this study, we investigated the impact of root exudates from compatible host plant species; Elaeagnus angustifolia on the exoproteome of Parafrankia soli strain NRRL B-16219. A total of 565 proteins were evidenced as differentially abundant, with 32 upregulated and 533 downregulated in presence of the plant exudates. Analysis of the function of these proteins suggests that the bacterial strain is undergoing a complex metabolic reprogramming towards a new developmental phase elicited in presence of host plant root exudates. The upregulation of Type II/IV secretion system proteins among the differentially expressed proteins indicates their possible role in infecting the host plant, as shown for some rhizobia. Additionally, EF-Tu, proteins upregulated in this study, may function as an effector for the T4SSs and trigger plant defense responses. These findings suggest that Parafrankia soli may use EF-Tu to infect the actinorhizal host plant and pave the way for further investigations of the molecular mechanisms underlying the establishment of symbiotic relationships.
Introduction
Parafrankia is a bacterial genus classified within the Frankiaceae family, alongside the genera Frankia, Protofrankia, and Pseudofrankia [1, 2]. Parafrankia strains are known for their ability to form symbiotic relationships with actinorhizal plants of the Elaeagnaceae, Colletieae (Rhamnaceae), Morella (Myricaceae), and Gynmnostoma (Casuarinaceae) species. Within the formed root nodules, the bacteria are able to fix atmospheric nitrogen, which can then be utilized by the plant as a nutrient source. The rhizosphere serves as the site for the symbiotic signalling cascade, which coordinates the regulation of genes and exchange of symbiotic signals [3]. This intricate process leads to mutual recognition and, ultimately, the formation of functional root nodules. Although actinorhizal and legume root nodules share many developmental characteristics [4], there are notable differences in certain molecular signals.
In legume symbiosis, flavonoids have been identified as crucial signalling molecules during the early stages [5]. These compounds act as chemotactic signals for rhizobia and specifically bind to the rhizobial NodD protein. As a result, this protein activates the transcription of nodulation genes essential for the synthesis of lipochito-oligosaccharide (LCO) Nod factors [6, 7]. Subsequently, these Nod factors transmit signals back to the host plant by binding to LysM receptor kinases, initiating the activation of the common symbiotic signalling pathway (CSSP). The CSSP is a shared signalling pathway found in both legume symbiosis and mycorrhizal symbiosis [8]. While there is a belief that actinorhizal plants also employ flavonoids as signalling molecules, there is currently a lack of direct evidence to support their role in the process [9-14]. In almost all frankia genomes, canonical nod genes are generally absent [15, 16]. Furthermore, there is no indication that early frankia signalling relies on canonical nodABC genes or molecules associated with rhizobial Nod factors, even when these genes are present in their genomes [17, 18]. The conserved symbiotic signalling pathway (CSSP) is also involved in the communication actinorhizal plant and their frankia microsymbionts [19, 20]. Typically, receptor complexes with LysM motifs are responsible for binding GlcNAc-based elicitors such as chitin, chitin oligosaccharides with lipid modifications; Myc factors [21] and Nod factors [22], and peptidoglycan [23-25] which consist of alternating GlcNAc and N-acetylmuramic acid residues linked by peptides. LysM-receptor-like kinases can also detect proteinaceous elicitors like flg22 from flagellin and nlp20 [26-28], a conserved epitope found in bacteria, fungi, and oomycetes [29, 30]. There are other receptor classes involved in recognizing lipopolysaccharides (lectin-like) [30]. Secretion systems have been demonstrated to play crucial metabolic roles in exporting various molecules including effectors which are instrumental in manipulating host cellular processes and also in sensing and responding to changes in the environment, particularly within the context of plant symbiotic bacteria [31, 32].
Studies have explored the cellular proteomes of Frankia alni, Protofrankia coriariae, and Parafrankia soli species following treatments with host root exudates in order to describe the induced molecular dynamics [18, 33, 34]. The results of these studies indicate that the symbiotic signalling systems in actinorhizal symbiosis are highly intricate and tightly regulated. According to a study conducted by Gueddou et al. [18], proteins involved in various biological processes showed increased expression when exposed to root exudates from Elaeagnus angustifolia. These proteins are associated with nitrogen fixation and assimilation, respiration, oxidative stress, proteolysis, and plant cell wall degradation. Thus far, there have been no significant findings of any candidate proteins linked to nodulation factors that can be sensed by LysM-receptor-like kinases and leading ultimately to a signal transduction cascade in actinorhizal plants.
The bacterial exoproteome is the entirety of proteins that a bacterial cell releases into the environment through secretion systems, outer membrane vesicles, or lysis [35; 36]. The proteins within the bacterial exoproteome have diverse functions in bacterial physiology [36-39]. This can include proteins that aid in nutrient acquisition, such as transporters and enzymes that break down complex molecules [40]. Additionally, the exoproteome can contain proteins that bind to host or microbial receptors, which allows for the mediation of signalling [41; 42]. Quorum sensing molecules are also present in the exoproteome and allow bacteria to coordinate their behaviour based on population density [39; 43; 44]. Some proteins within the exoproteome can also modulate the host immune system, suppressing host defences [39; 45] or promoting the growth of host tissues [46-48].
In the early stages following plant stimuli, it has been shown that rhizobial exoproteome comprises adhesins that assist in bacterial attachment to roots, enzymes necessary for the modification of surface polysaccharides, and effectors that can either suppress plant defense responses or activate specific signalling pathways [7].
In this study, we present the extracellular proteome analysis of Parafrankia soli strain NRRL B-16219, which was treated with root exudates in a minimal medium. The goal was to identify whether the secreted proteins during the early response phase of Parafrankia soli strain NRRL B-16219 to plant stimuli contained a significant amount of symbiotically relevant proteins and could provide insights into symbiotic signalling.
Materials and Methods
Production of root exudates
To obtain root exudates, Elaeagnus angustifolia seedlings were grown axenically in Broughton and Dilworth [49] nutrient solution supplemented with 5 mM KNO3 as the nitrogen source (BD+N). Two weeks later, the BD+N medium was replaced with nitrogen-free BD medium (BD-N and root exudates were collected after two additional weeks seedling growth. The collected exudates underwent filter sterilization using a 0.22 µm polycarbonate membrane.
Bacterial growth conditions and protein extraction
Parafrankia soli strain NRRL B-16219 [50] was cultivated in 125 ml bottles containing 40 ml of Broughton and Dilworth solution without nitrogen (BD-N), supplemented with 5 mM pyruvate as the carbon source, at a temperature of 28°C without shaking. After five days of exponential growth, one volume (v/v) of freshly collected root exudate was introduced to the culture. In control experiments, one volume of BD-N was added to NRRL B-16219 cultures grown under identical conditions. Following three days of exposure to the root exudates, the exoproteomes of NRRL B-16219 were analysed using the methods described previously [34]. Each experiment consisted of four independent biological replicates.
Nano-liquid chromatography and tandem mass spectrometry analysis
To analyse the peptide digests, we employed an Ultimate 3000 LC system (Thermo-Scientific, Villebon-sur-Yvette, France), following the detailed protocol outlined in Ktari et al. [34] and subsequently in Gueddou et al. [18]. The MS/MS spectra were examined using the MASCOT 2.3.02 search engine (Matrix Science, London, UK) with standard parameters, as described by Hartmann and Armengaud (2014). The search was conducted against the complete list of annotated CDS from the draft genome of Parafrankia soli strain NRRL B-16219 (GenBank/EMBL/DDBJ accession number MAXA00000000.1), which comprises 6,679 protein sequences [17]. Peptide matches exceeding the peptidic identity threshold were filtered based on a significance level of P < 0.05. Validated proteins were those that had at least two peptide sequences assigned to them, following the principle of parsimony. For protein abundance evaluation, we employed a previously described approach [51; 52] involving shotgun analysis with MS/MS spectral counts. The calculation of normalized spectral count abundance factors was performed following the methodology outlined by Paoletti et al. [53]. The resulting values were expressed as percentages of the total signal.
Data analysis
Computational predictions of protein subcellular localization data were performed based on Subcellular localization of proteins was predicted with PrediSi software [54]. Signal peptide sequences were further investigated at the CBS prediction server (http://www.cbs.dtu.dk./services/), using SignalP version 6.0 [55], TatP version 1.0 [56], and SecretomeP version 2.0 [57].
Type IV secretion system proteins were identified by T4SEpre (beta) [58] which predicts Type IV secreted proteins based on amino acid composition in C-termini and using EffectiveDB [59] with the plant classification module and selective (0.5) restriction value method enabled.
Differentially detected proteins were categorized into functional classes and re-annotated using FUNAGE-Pro v1 software [60]. FUNAGE-Pro also allows enrichment analysis and additionally predicts most relevant functions.
Results
General characteristics of Parafrankia soli NRRL B-16219 exoproteome
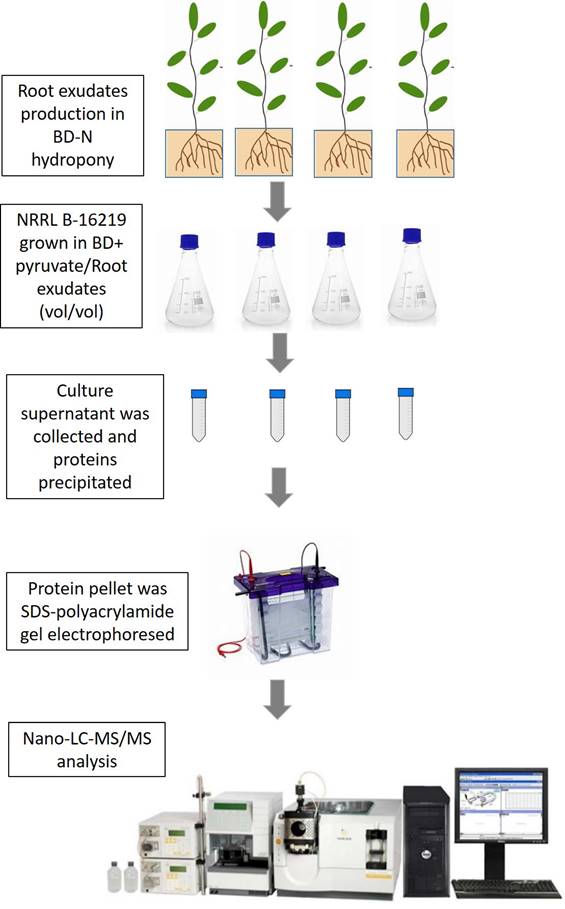
The experimental pipeline for producing root exudates, treat NRRL B-16219, and analysing the exoproteome by next-generation shotgun proteomics using high resolution nanoLC-MS/MS, was summarized in Fig. 1. As expected, the root exudate by itself was very low in terms of protein load and resulted in negligible peptide identification [18]. The analysis of the exoproteome from strain NRRL B-16219 using tandem mass spectrometry generated a total of 306,055 MS/MS spectra, among which 39,216 could be confidently assigned to peptide sequences when results from all samples were combined (Supplementary Tables S1). The percentage of assignment reached 29% for the exoproteome of untreated bacteria, a ratio commonly found for other bacteria [61; 62]. The experimental dataset comprised a total of 7,324 peptide sequences that were attributed to 2,011 proteins detected, with 948 of them certified by at least 2 peptides, representing the exoproteome from strain NRRL B-16219 grown in the presence or absence of root exudates. Additional information such as the abundance of each of these proteins per replicate and condition can be found in Supplementary Table S2-S3.
Experimental pipeline used in the present study for root exudate production, treatment of NRRL B-16219, protein precipitation and NanoLC-MS/MS analysis.
A threshold of ± fold (≥1.5) with a p-value ≤0.05 was employed to identify proteins that were differentially detected, either up- or down-regulated. Out of the 565 differential abundant proteins in the presence of E. angustifolia root exudates, there were 32 upregulated and 533 downregulated proteins (Supplementary table S4).
Most of differentially abundant proteins were predicted to be cytoplasmic (53.6%) followed by unknown localisation (24.2%) and membrane/cytoplasmic (17.7%). Fewer were predicted to occur in the extracellular (2.4%) or cell wall (2.1%) compartments (Supplementary Table S3).
Result for the detection of signal peptide sequences with potential cleavage site was found to be thin. Exception is for “MULTISPECIES_ substrate-binding domain-containing protein” (WP_083390861.1) putatively secreted through Sec machinery with signal peptide probability of 0.78 and probable cleavage site between 43-44 residues. The “Aspartate aminotransferase family protein” (WP_071066848.1) was predicted to route through twin-arginine translocation (Tat) pathway with a cleavage site most likely between position 46 and 47 residues.
Functional analysis of differently expressed proteins
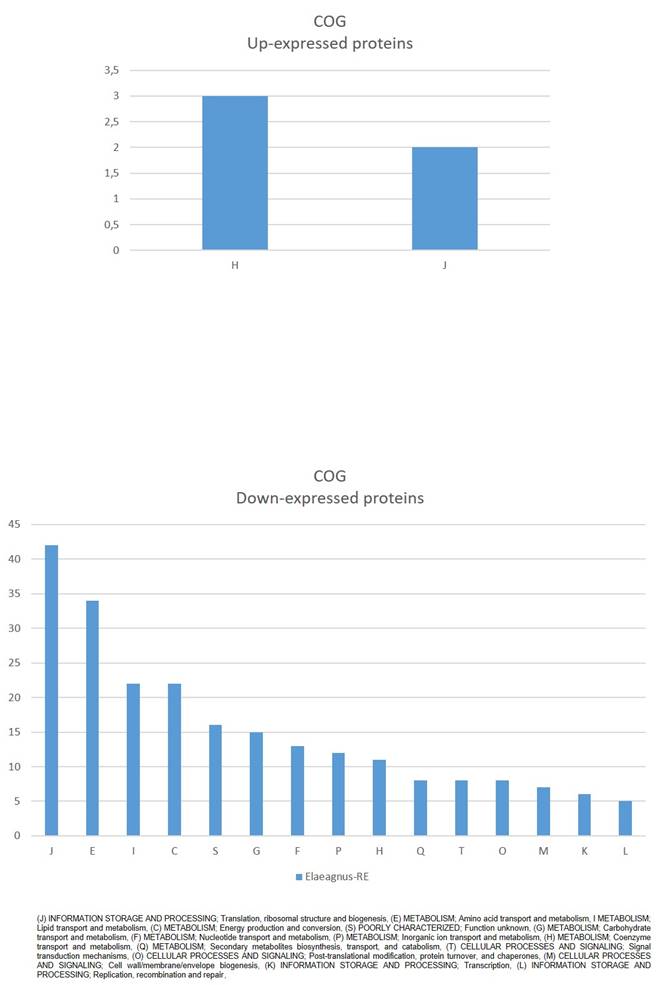
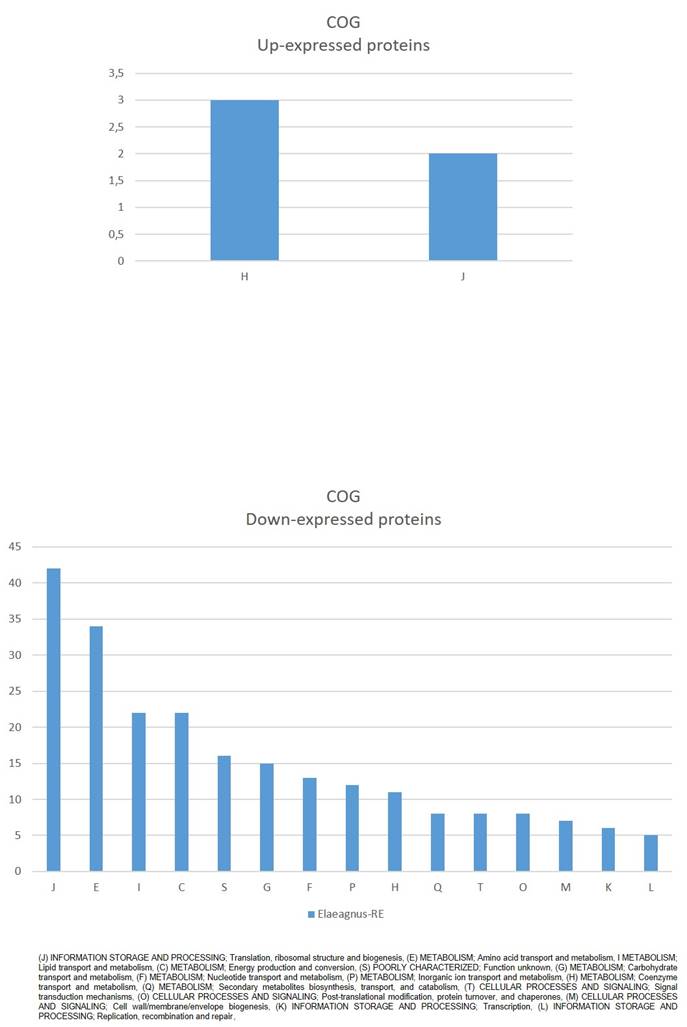
Clusters of Orthologous Groups (COG) mapping (Fig. 2) showed that down-expressed proteins were mostly (J) INFORMATION STORAGE AND PROCESSING; Translation, ribosomal structure and biogenesis, (E) METABOLISM; Amino acid transport and metabolism, I METABOLISM; Lipid transport and metabolism, (C) METABOLISM; Energy production and conversion, among others. While up-expressed proteins were assigned to (H) METABOLISM; Coenzyme transport and metabolism, and (J) INFORMATION STORAGE AND PROCESSING; Translation, ribosomal structure and biogenesis (Fig. 2).
COG classification of significantly affected proteins in Parafrankia soli strain NRRL B-16219 exoproteome in presence of root exudates of E. angustifolia host species. A graphical representation of up- (a) and down-expressed proteins (p value ≤ 0.05, Tfold ≥ 1.5 and hits/class score = 9 (0-9, 0 being not significant, ranked based on Benjamini-Hochberg Algorithm)).
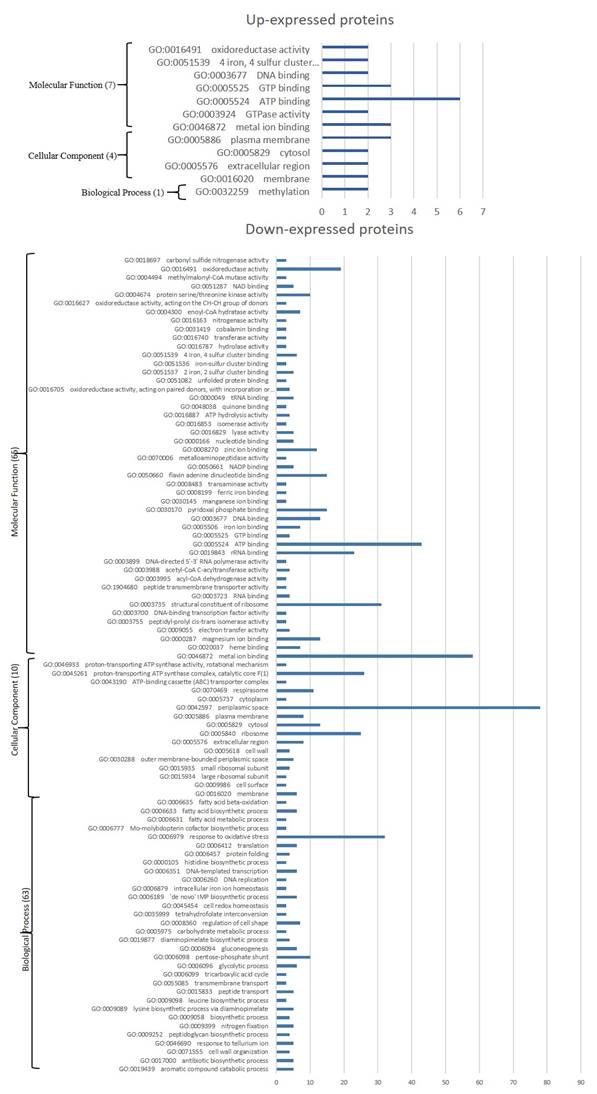
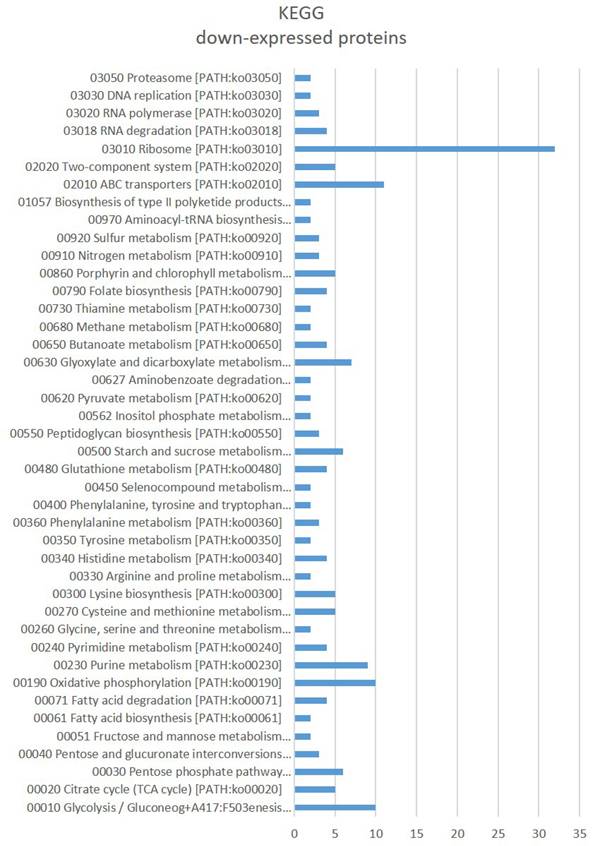
Gene Ontology (GO) enrichment (Fig. 3) assigned dawn-expressed proteins mainly to “metal ion binding”, “ATP binding”, 'structural constituent of ribosome”, “RNA binding”, 'oxidoreductase activity' among others within Molecular Function, “cytoplasm”, “plasma membrane” and 'ribosome' among others as Cellular Component, and Biological Process include “translation” and “tricarboxylic acid cycle” among others. Kyoto Encyclopedia of Genes and Genomes (KEGG) showed “Ribosome”, ABC transporters, Glycolysis / Gluconeogenesis, “Oxidative phosphorylation”, “Purine metabolism”, “Glyoxylate and dicarboxylate metabolism”, “Starch and sucrose metabolism” and 'Pentose phosphate pathway” amongst the main identified sets (Fig. 4).
GO terms significantly affected in Parafrankia soli strain NRRL B-16219 exoproteome in presence of root exudates of E. angustifolia host species. A graphical representation of up- (a) and down-expressed proteins (p value ≤ 0.05, Tfold ≥ 1.5 and hits/class score = 9 (0-9, 0 being not significant, ranked based on Benjamini-Hochberg Algorithm)). Only hits/class size >2 are presented.
KEGG terms significantly affected in Parafrankia soli strain NRRL B-16219 exoproteome in presence of root exudates of E. angustifolia host species. A graphical representation of down-expressed proteins (p value ≤ 0.05, Tfold ≥ 1.5 and hits/class score = 9 (0-9, 0 being not significant, ranked based on Benjamini-Hochberg Algorithm)).
GO enrichment of up-expressed proteins (Fig. 2) mainly identify “ATP binding', 'GTP binding' and “metal ion binding” among others for Molecular Function, “plasma membrane” for Cellular Component among others, and “methylation' Biological Process. Among up-expressed proteins only “Cysteine and methionine metabolism” was identified as connected to KEGG pathway.
To gain insight functional analysis up-regulated proteins were further assigned based on InterPro functional classification (Table 1). Interestingly “S-adenosyl-L-methionine-dependent methyltransferase”, “Type II/IV secretion system protein” and “Elongation factor Tu” were identified. The latter “Elongation factor Tu” was the only predicted as Type IV secretion system's effector among up-expressed proteins.
Statistically significant up-expressed proteins (p value ≤ 0.05 and a Tfold ≥ 1.5) categorized into functional classes and annotated by COG, GO, KEGG and InterPro analysis using FUNAGE-Pro v1 software (de Jong et al., 2022)
| Class ID | Description | Score; Hits/Class Size; p-value* | |
|---|---|---|---|
| IPR000640 | Elongation factor EFG, domain V-like | 9; 2/2; 0.00 | WP_071059306.1,WP_071066147.1 |
| IPR029058 | Alpha/Beta hydrolase fold | 9; 2/2; 0.00 | WP_071063096.1,WP_071066904.1 |
| GO:0005524 | ATP binding | 9; 6/6; 0.00 | WP_071059701.1,WP_071064601.1,WP_071064902.1,WP_071065597.1,WP_071066117.1,WP_071066193.1 |
| IPR027417 | P-loop containing nucleoside triphosphate hydrolase | 9; 7/7; 0.00 | WP_071059306.1,WP_071059701.1,WP_071064601.1,WP_071064902.1,WP_071065597.1,WP_071066147.1,WP_071066193.1 |
| 00270 Cysteine and methionine metabolism [PATH:ko00270] | ko00270 | 9; 2/2; 0.00 | WP_071060170.1,WP_071064376.1 |
| IPR031157 | Tr-type G domain, conserved site | 9; 2/2; 0.00 | WP_071059306.1,WP_071066147.1 |
| J | INFORMATION STORAGE AND PROCESSING; Translation, ribosomal structure and biogenesis | 9; 2/2; 0.00 | WP_071064582.1,WP_071066147.1 |
| IPR009000 | Translation protein, beta-barrel domain superfamily | 9; 2/2; 0.00 | WP_071059306.1,WP_071066147.1 |
| IPR041095 | Elongation Factor G, domain II | 9; 2/2; 0.00 | WP_071059306.1,WP_071066147.1 |
| IPR000795 | Transcription factor, GTP-binding domain | 9; 2/2; 0.00 | WP_071059306.1,WP_071066147.1 |
| IPR036188 | FAD/NAD(P)-binding domain superfamily | 9; 2/2; 0.00 | WP_071059950.1,WP_071066784.1 |
| IPR022399 | Helicase/secretion neighbourhood ATPase | 9; 2/2; 0.00 | WP_071064601.1,WP_071066193.1 |
| IPR035647 | EF-G domain III/V-like | 9; 2/2; 0.00 | WP_071059306.1,WP_071066147.1 |
| GO:0005525 | GTP binding | 9; 3/3; 0.00 | WP_071059306.1,WP_071066147.1,WP_083390659.1 |
| GO:0032259 | methylation | 9; 2/2; 0.00 | WP_071060170.1,WP_071066904.1 |
| GO:0051539 | 4 iron, 4 sulfur cluster binding | 9; 2/2; 0.00 | WP_071060541.1,WP_083390659.1 |
| GO:0003677 | DNA binding | 9; 2/2; 0.00 | WP_071062294.1,WP_071065581.1 |
| IPR029063 | S-adenosyl-L-methionine-dependent methyltransferase | 9; 2/2; 0.00 | WP_071065581.1,WP_071066904.1 |
| GO:0046872 | metal ion binding | 9; 3/3; 0.00 | WP_071060541.1,WP_071066896.1,WP_083390659.1 |
| IPR005225 | Small GTP-binding protein domain | 9; 2/2; 0.00 | WP_071059306.1,WP_071066147.1 |
| GO:0005576 | extracellular region | 9; 2/2; 0.00 | WP_071060541.1,WP_071066896.1 |
| GO:0005829 | cytosol | 9; 2/2; 0.00 | WP_071060170.1,WP_071060541.1 |
| GO:0005886 | plasma membrane | 9; 3/3; 0.00 | WP_071060541.1,WP_071064376.1,WP_071067042.1 |
| GO:0016491 | oxidoreductase activity | 9; 2/2; 0.00 | WP_071059950.1,WP_071066784.1 |
| GO:0003924 | GTPase activity | 9; 2/2; 0.00 | WP_071059306.1,WP_071066147.1 |
| GO:0016021 | integral component of membrane | 9; 2/2; 0.00 | WP_071064376.1,WP_071067042.1 |
| GO:0000746 | conjugation | 9; 2/2; 0.00 | WP_071064601.1,WP_071066193.1 |
| H | METABOLISM; Coenzyme transport and metabolism | 9; 3/3; 0.00 | WP_020463537.1,WP_071066904.1,WP_083390659.1 |
| IPR004161 | Translation elongation factor EFTu-like, domain 2 | 9; 2/2; 0.00 | WP_071059306.1,WP_071066147.1 |
| IPR001482 | Type II/IV secretion system protein | 9; 2/2; 0.00 | WP_071064601.1,WP_071066193.1 |
| Enzymes | Oxidoreductases ndh; NADH:quinone reductase (non-electrogenic) [EC:1.6.5.9] Transferases metH, MTR; 5-methyltetrahydrofolate--homocysteine methyltransferase [EC:2.1.1.13], NMT; phosphoethanolamine N-methyltransferase [EC:2.1.1.103] small RNA 2'-O-methyltransferase [EC:2.1.1.386] pks12; mycoketide-CoA synthase [EC:2.3.1.295] ubiX, bsdB, PAD1; flavin prenyltransferase [EC:2.5.1.129] mucR; diguanylate cyclase [EC:2.7.7.65] Hydrolases atzF; allophanate hydrolase [EC:3.5.1.54] hrpB; ATP-dependent helicase HrpB [EC:3.6.4.13] Lyases DDC, TDC; aromatic-L-amino-acid/L-tryptophan decarboxylase [EC:4.1.1.28 4.1.1.105] moaA, CNX2; GTP 3',8-cyclase [EC:4.1.99.22] ACO, acnA; aconitate hydratase [EC:4.2.1.3] Isomerases groEL, HSPD1; chaperonin GroEL [EC:5.6.1.7] Translocases cpaF, tadA; pilus assembly protein CpaF [EC:7.4.2.8] | WP_071066784.1 WP_071060170.1 WP_071066904.1 WP_071064742.1 WP_071063096.1 WP_020463537.1 WP_071067042.1 WP_071060056.1 WP_071064902.1 WP_071066848.1 WP_083390659.1 WP_071060541.1 WP_071066117.1 WP_071066193.1, WP_071064601.1 |
*The values correspond to Score (0-9, 0 being not significant, ranked based on Benjamini-Hochberg Algorithm), p value and the hits/class size. Not applicable for enzymes EC assignment.
Detected enzymes are mainly Transferases (5), followed by Lyases (3), Hydrolases (2), and Oxydoreductases, Isomerases and Translocases (1 each) as indicated in Table 1.
Discussion
The abundance exoproteins, detected in NRRL B-16219 without root exudate treatment, including a high proportion of proteins predicted to have cellular localisation, may be explained by extensive cell autolysis which is a common feature of microbial growth associated with a variety of cultural factors and stresses (Shockman et al., 1996). For frankia, studies have reported a similar trend of autolysis in both static and stirred defined medium [64-66], under nitrogen-fixing conditions [67], and even in aging nodules [68]. Mastronunzio et al. [69] suggested that detected proteins in exoproteome is due to frankia cell lysis during growth rather than true secretion. There is a possibility that more secreted proteins in Frankia are present, but they might be either attached to the membrane or associated with the cell envelope, thus not being detected in the medium. In the present study the exoproteome of strain NRRL B-16219 showed a significant decrease in abundance after five days of growth when exposed to root exudates compared to the control condition. This observation suggests that the cellular autolysis may ceased indicating a potential recovery of growth. Most studies attributed carbon source depletion as a stressful condition that can trigger cellular proteolysis, which in turn can lead to autolysis [70]. While in rich organic media most analysed frankia displayed long stationary phases and cells remain viable up to one year [65]. Therefore, NRRL B-16219 may perceive root exudates as abundant nutrient sources, potentially postponing its cell autolysis and promoting the continuous exponential growth of the strain.
Proteins involved in autolysis consisted of proteasomes (WP_071063554.1, WP_071063556.1 and WP_071063560.1), aminopeptidase (WP_071065370.1, WP_071061455.1, WP_071059318.1, WP_071066137.1, WP_071063686.1), and peptidoglycan endopeptidase (WP_071063732.1) which may be seen as responsible for catabolism of the cell-wall under nutrient deficiency [71]. Benoist et al. (1992) [70] reported that these proteinase subunits exhibited a significant increase in activity upon cessation of growth within 5 days old culture in stirred mineral medium. The addition of fresh BAP medium or carbon source (Propionate), but not nitrogen source (NH4Cl), at the end of the exponential growth phase extended growth for an additional day and delayed the increase in activity of the proteinase subunits for 3 days after cessation of growth. However, upon resuspending frankia cells in the late exponential phase (3 days) in a culture filtrate obtained from a 5-day-old culture and supplemented with BAP-PCM medium components, the biomass yield decreased to approximately 50%.
Another downregulated protein thought to be related to autolysis is GlcNAc-PI de-N-acetylase (WP_020458433.1), which is involved in the hydrolysis of the bacterial cell wall and signalling the need for development under nutrient-limiting conditions. According to van Bergeijk et al. [72], autolytic degradation of the cell-wall peptidoglycan releases amino sugars such as GlcNAc and N-acetylmuramic acid (MurNAc) around the colonies. GlcNAc accumulation triggers development and antibiotic production under famine conditions (signalling starvation), while it blocks both processes under feast conditions (signalling abundance) [73]. Another novel concept is advanced that peptidoglycan deacylases is proposed to be seen as virulence factors [74; 75].
Among the up-expressed proteins S-adenosyl-L-methionine-dependent methyltransferase (WP_071065581.1, WP_071066904.1). Geelen et al. [76] suggested that NodS acts as a methyltransferase that depends on S-adenosyl-L-methionine and is necessary for the methylation of chitin oligosaccharides lacking acetyl groups at the non-reducing end. Because no upstream Nod proteins of NRRL B-16219 were detected in its cellular proteome [18] nor, here, in its exoproteome, this protein may act differently to what has been described for rhizobia signalling.
Bacterial type IV secretion systems (T4SSs) (WP_071064601.1, WP_071066193.1), which are up-expressed proteins, belong to the bacterial type IV secretion systems (T4SSs). This is a diverse translocation superfamily, as noted by Grohmann et al. [77], that encompasses various functions. The T4SSs are primarily composed of two major subfamilies: (i) conjugative systems that enable transfer of DNA between bacteria, and (ii) effector translocators that either inject effector macromolecules directly into prokaryotic or eukaryotic host cells or secrete them into the surrounding medium, leading to a variety of effects on host cell functions during infection [78].
It has been shown that Mesorhizobium loti and Sinirhizobium meliloti secrete via a T4SSs specific proteins that affect nodulation [79; 80]. Notably, in certain rhizobia, the genes involved in nodulation factor synthesis and encoding the type IV secretion system are under the control of a common regulator that is activated by flavonoids released by root legumes [6].
Up-expressed proteins EF-Tu (WP_071059306.1, WP_071066147.1) were the only predicted in this study as effector of T4SS. EF-Tu have been detected in the exoproteomes of many microbial pathogens [81-84], where it affects calcium cycling and elicits plant defense responses such as promoting Ca2+ influx across the membrane, induction of an oxidative burst, activation of calcium-dependent protein kinases and mitogen-activated protein kinase (MAPK) cascades [81; 85]. Most of these plant reactions have been observed in root epidermal cells following the infection by rhizobia [86] or frankia [87].
In conclusion, the observed alterations of the exoproteome of Parafrankia soli strain NRRL B-16219 in response to root exudates of E. angustifolia indicates that the strain is adapting to its new surroundings microniche. The differential expression of proteins indicates that the strain might be undergoing a complex metabolic reprogramming and ceasing autolysis to acquire and utilize nutrients for a new developmental phase as part of the transition on the road to symbiotic lifestyle. The identification of T4SSs among the up-expressed proteins suggests that they may play a crucial role in infecting the host plant, similar to some rhizobia which use T4SSs to positively or negatively influence nodulation. Additionally, EF-Tu, which was up-regulated in this study, could serve as an effector for the identified T4SSs. EF-Tu has been detected in numerous microbial pathogens and symbionts and has been shown to trigger plant defense responses, particularly in root epidermal cells. Hence, it is plausible that frankia employs EF-Tu effector during actinorhizal plant infection. The up-regulated S-adenosyl-L-methionine-dependent methyltransferase in NRRL B-16219, without detectable upstream Nod proteins, suggests a distinct signalling role from rhizobial nod-dependent pathways.
Supplementary Material
Supplementary tables.
Acknowledgements
Funding
This study was supported by MESRS funding to the Unité de Bacteriologie Moléculaire & Génomique, Institut National des Sciences Appliquées et de Technologie, Université de Carthage, Tunisia.
Data availability statement
The data underlying this article are available in the article and in its online supplementary material.
Author contributions
MG conceived of the project, IK and FGG carried out plant and bacterial experiments, GM and JA carried out the proteomic analysis, MG analysed the data and wrote the manuscript. All of the authors approved the submitted manuscript.
Ethical approval
This article does not include any studies involving human participants or animals.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gtari M. Taxogenomic status of phylogenetically distant Frankia clusters warrants their elevation to the rank of genus: A description of Protofrankia gen. nov, Parafrankia gen. nov, and Pseudofrankia gen. nov. as three novel genera within the family Frankiaceae. Front Microbiol. 2022;13:1041425
2. Oren A, Göker M. Validation List no. 210. Valid publication of new names and new combinations effectively published outside the IJSEM. Int J Syst Evol Microbiol. 2023;73(3):1-6
3. Venturi V, Keel C. Signaling in the rhizosphere. Trends Plant Sci. 2016;21(3):187-198
4. Pawlowski K, Bisseling T. Rhizobial and actinorhizal symbioses: what are the shared features? Plant Cell. 1996;8(10):1899
5. Brewin NJ. Plant cell wall remodelling in the Rhizobium-legume symbiosis. Crit Rev Plant Sci. 2004;23(4):293-316
6. Deakin WJ, Broughton WJ. Symbiotic use of pathogenic strategies: rhizobial protein secretion systems. Nat Rev Microbiol. 2009;7(4):312-320
7. Downie JA. The roles of extracellular proteins, polysaccharides and signals in the interactions of rhizobia with legume roots. FEMS Microbiol Rev. 2010;34(2):150-170
8. Oldroyd GE. Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nat Rev Microbiol. 2013;11(4):252-263
9. Hocher V, Ngom M, Carré-Mlouka A, Tisseyre P, Gherbi H, Svistoonoff S. Signalling in actinorhizal root nodule symbioses. Antonie Van Leeuwenhoek. 2019;112(1):23-29
10. Auguy F, Abdel-Lateif K, Doumas P, Badin P, Guerin V, Bogusz D, Hocher V. Activation of the isoflavonoid pathway in actinorhizal symbioses. Funct Plant Biol. 2011;38(9):690-696
11. Abdel-Lateif K, Bogusz D, Hocher V. The role of flavonoids in the establishment of plant roots endosymbioses with arbuscular mycorrhiza fungi, rhizobia and Frankia bacteria. Plant Signal Behav. 2012;7(6):636-641
12. Popovici J, Comte G, Bagnarol É, Alloisio N, Fournier P, Bellvert F. et al. Differential effects of rare specific flavonoids on compatible and incompatible strains in the Myrica gale-Frankia actinorhizal symbiosis. Appl Environ Microbiol. 2010;76(8):2451-2460
13. Popovici J, Walker V, Bertrand C, Bellvert F, Fernandez MP, Comte G. Strain specificity in the Myricaceae-Frankia symbiosis is correlated to plant root phenolics. Funct Plant Biol. 2011;38(9):682-689
14. Benoit LF, Berry AM. Flavonoid-like compounds from seeds of red alder (Alnus rubra) influence host nodulation by Frankia (Actinomycetales). Physiol Plant. 1997;99(4):588-593
15. Normand P, Lapierre P, Tisa LS, Gogarten JP, Alloisio N, Bagnarol E. et al. Genome characteristics of facultatively symbiotic Frankia sp. strains reflect host range and host plant biogeography. Genome Res. 2007;17(1):7-15
16. Tisa LS, Oshone R, Sarkar I, Ktari A, Sen A, Gtari M. Genomic approaches toward understanding the actinorhizal symbiosis: an update on the status of the Frankia genomes. Symbiosis. 2016;70:5-16
17. Ktari A, Nouioui I, Furnholm T, Swanson E, Ghodhbane-Gtari F, Tisa LS, Gtari M. Permanent draft genome sequence of Frankia sp. NRRL B-16219 reveals the presence of canonical nod genes, which are highly homologous to those detected in Candidatus Frankia Dg1 genome. Stand Genomic Sci. 2017b;12(1):1-10.
18. Gueddou A, Sarker I, Sen A, Ghodhbane-Gtari F, Benson DR, Armengaud J, Gtari M. Effect of actinorhizal root exudates on the proteomes of Frankia soli NRRL B-16219, a strain colonizing the root tissues of its actinorhizal host via intercellular pathway. Res Microbiol. 2022;173(1-2):103900
19. Gherbi H, Markmann K, Svistoonoff S, Estevan J, Autran D, Giczey G. et al. SymRK defines a common genetic basis for plant root endosymbioses with arbuscular mycorrhiza fungi, rhizobia, and Frankia bacteria. Proc Natl Acad Sci. 2008;105(12):4928-4932
20. Hocher V, Alloisio N, Auguy F, Fournier P, Doumas P, Pujic P. et al. Transcriptomics of actinorhizal symbioses reveals homologs of the whole common symbiotic signaling cascade. Plant Physiol. 2011;156(2):700-711
21. Maillet F, Poinsot V, André O, Puech-Pagès V, Haouy A, Gueunier M. et al. Fungal lipochitooligosaccharide symbiotic signals in arbuscular mycorrhiza. Nature. 2011;469(7328):58-63
22. Oldroyd GE. Dissecting symbiosis: developments in Nod factor signal transduction. Ann Bot. 2001;87(6):709-718
23. Willmann R, Lajunen HM, Erbs G, Newman MA, Kolb D, Tsuda K, Nürnberger T. Arabidopsis lysin-motif proteins LYM1 LYM3 CERK1 mediate bacterial peptidoglycan sensing and immunity to bacterial infection. Proc Natl Acad Sci. 2011;108(49):19824-19829
24. Kouzai Y, Mochizuki S, Nakajima K, Desaki Y, Hayafune M, Miyazaki H. et al. Targeted gene disruption of OsCERK1 reveals its indispensable role in chitin perception and involvement in the peptidoglycan response and immunity in rice. Mol Plant Microbe Interact. 2014;27(9):975-982
25. Ao Y, Li Z, Feng D, Xiong F, Liu J, Li JF. et al. Os CERK 1 and Os RLCK 176 play important roles in peptidoglycan and chitin signaling in rice innate immunity. Plant J. 2014;80(6):1072-1084
26. Lopez-Gomez M, Sandal N, Stougaard J, Boller T. Interplay of flg22-induced defence responses and nodulation in Lotus japonicus. J Exp Bot. 2012;63(1):393-401
27. Thor K, Peiter E. Cytosolic calcium signals elicited by the pathogen-associated molecular pattern flg22 in stomatal guard cells are of an oscillatory nature. New Phytol. 2014;204(4):873-881
28. Keinath NF, Waadt R, Brugman R, Schroeder JI, Grossmann G, Schumacher K, Krebs M. Live cell imaging with R-GECO1 sheds light on flg22-and chitin-induced transient [Ca2+] cyt patterns in Arabidopsis. Mol Plant. 2015;8(8):1188-1200
29. Albert I, Zhang L, Bemm H, Nürnberger T. Structure-function analysis of immune receptor at RLP23 with its ligand nlp20 and coreceptors at SOBIR1 and At BAK1. Mol Plant Microbe Interact. 2019;32(8):1038-1046
30. Zipfel C, Oldroyd GE. Plant signalling in symbiosis and immunity. Nature. 2017;543(7645):328-336
31. Nelson MS, Sadowsky MJ. Secretion systems and signal exchange between nitrogen-fixing rhizobia and legumes. Front Plant Sci. 2015;6:491
32. Lucke M, Correa MG, Levy A. The role of secretion systems, effectors, and secondary metabolites of beneficial rhizobacteria in interactions with plants and microbes. Front Plant Sci. 2020;11:589416
33. Pujic P, Alloisio N, Fournier P, Roche D, Sghaier H, Miotello G. et al. Omics of the early molecular dialogue between Frankia alni and Alnus glutinosa and the cellulase synton. Environ Microbiol. 2019;21(9):3328-3345
34. Ktari A, Gueddou A, Nouioui I, Miotello G, Sarkar I, Ghodhbane-Gtari F. et al. Host plant compatibility shapes the proteogenome of Frankia coriariae. Front Microbiol. 2017;8:720
35. Wang G, Chen H, Xia Y, Cui J, Gu Z, Song Y. et al. How are the non-classically secreted bacterial proteins released into the extracellular milieu? Curr Microbiol. 2013;67:688-695
36. Armengaud J, Christie-Oleza JA, Clair G, Malard V, Duport C. Exoproteomics: exploring the world around biological systems. Expert Rev Proteomics. 2012;9(5):561-575
37. Desvaux M, Hébraud M, Talon R, Henderson IR. Secretion and subcellular localizations of bacterial proteins: a semantic awareness issue. Trends Microbiol. 2009;17(4):139-145
38. Maffei B, Francetic O, Subtil A. Tracking proteins secreted by bacteria: what's in the toolbox? Front Cell Infect Microbiol. 2017;7:221
39. Sauvage S, Hardouin J. Exoproteomics for better understanding Pseudomonas aeruginosa virulence. Toxins. 2020;12(9):571
40. von Tils D, Blädel I, Schmidt MA, Heusipp G. Type II secretion in Yersinia—a secretion system for pathogenicity and environmental fitness. Front Cell Infect Microbiol. 2012;2:160
41. Cabrita P, Trigo MJ, Ferreira RB, Brito L. Is the exoproteome important for bacterial pathogenesis?. Lessons learned from interstrain exoprotein diversity in Listeria monocytogenes grown at different temperatures. Omics. 2014;18(9):553-569
42. Mulcahy ME, McLoughlin RM. Host-bacterial crosstalk determines Staphylococcus aureus nasal colonization. Trends Microbiol. 2016;24(11):872-886
43. Savinova OS, Glazunova OA, Moiseenko KV, Begunova AV, Rozhkova IV, Fedorova TV. Exoproteome analysis of antagonistic interactions between the probiotic bacteria Limosilactobacillus reuteri LR1 and Lacticaseibacillus rhamnosus F and multidrug resistant strain of Klebsiella pneumonia. Int J Mol Sci. 2021;22(20):10999
44. Ciprandi A, Da Silva WM, Santos AV, de Castro Pimenta AM, Carepo MSP, Schneider MPC. et al. Chromobacterium violaceum: important insights for virulence and biotechnological potential by exoproteomic studies. Curr Microbiol. 2013;67:100-106
45. Biagini M, Bagnoli F, Norais N. Surface and Exoproteomes of Gram-Positive Pathogens for Vaccine Discovery. In: Protein and Sugar Export and Assembly in Gram-positive Bacteria. 2017:309-337
46. Dou D, Zhou J-MM. Phytopathogen efectors subverting host immunity: Diferent foes, similar battleground. Cell Host Microbe. 2012;12:484-495
47. Khatabi B, Gharechahi J, Ghaffari MR, Liu D, Haynes PA, McKay MJ. et al. Plant-microbe symbiosis: what has proteomics taught us? Proteomics. 2019;19(16):1800105
48. Lidbury ID, Raguideau S, Liu S, Murphy AR, Stark R, Borsetto C. et al. Meta-exoproteomics identifies active plant-microbe interactions operating in the rhizosphere. bioRxiv. 2021-09.
49. Broughton WJ, Dilworth M. Control of leghaemoglobin synthesis in snake beans. Biochem J. 1971;125(4):1075-1080
50. Gtari M, Ghodhbane-Gtari F, Nouioui I. Frankia soli sp. nov, an actinobacterium isolated from soil beneath Ceanothus jepsonii. Int J Syst Evol Microbiol. 2020;70(2):1203-1209
51. Liu H, Sadygov RG, Yates JR. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193-4201
52. Zivanovic Y, Armengaud J, Lagorce A, Leplat C, Guérin P, Dutertre M. et al. Genome analysis and genome-wide proteomics of Thermococcus gammatolerans, the most radioresistant organism known amongst the Archaea. Genome Biol. 2009;10(6):1-23
53. Paoletti AC, Parmely TJ, Tomomori-Sato C, Sato S, Zhu D, Conaway RC. et al. Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc Natl Acad Sci. 2006;103(50):18928-18933
54. Hiller K, Grote A, Scheer M, Münch R, Jahn D. PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res. 2004;32(suppl_2):W375-W379
55. Teufel F, Almagro Armenteros JJ, Johansen AR, Gíslason MH, Pihl SI, Tsirigos KD. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol. 2022;40(7):1023-1025
56. Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S. Prediction of twin-arginine signal peptides. BMC Bioinformatics. 2005;6(1):1-9
57. Bendtsen JD, Kiemer L, Fausboll A, Brunak S. Non-classical protein secretion in bacteria. BMC Microbiol. 2005;5:58-70
58. Wang Y, Wei X, Bao H, Liu SL. Prediction of bacterial type IV secreted effectors by C-terminal features. BMC Genomics. 2014;15(1):1-14
59. Eichinger V, Nussbaumer T, Platzer A, Jehl MA, Arnold R, Rattei T. EffectiveDB—updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. 2016;44(D1):D669-D674
60. de Jong A, Kuipers OP, Kok J. FUNAGE-Pro: comprehensive web server for gene set enrichment analysis of prokaryotes. Nucleic Acids Res. 2022;50(W1):W330-W336
61. Rubiano-Labrador C, Bland C, Miotello G, Armengaud J, Baena S. Salt stress induced changes in the exoproteome of the halotolerant bacterium Tistlia consotensis deciphered by proteogenomics. PLoS One. 2015;10(8):e0135065
62. Madeira JP, Alpha-Bazin BM, Armengaud J, Duport C. Methionine residues in exoproteins and their recycling by methionine sulfoxide reductase AB serve as an antioxidant strategy in Bacillus cereus. Front Microbiol. 2017;8:1342
63. Shockman GD, Daneo-Moore L, Kariyama R, Massidda O. Bacterial walls, peptidoglycan hydrolases, autolysins, and autolysis. Microb Drug Resist. 1996;2(1):95-98
64. Lopez MF, Fontaine MS, Torrey JG. Levels of trehalose and glycogen in Frankia sp. HFPArI3 (Actinomycetales). Can J Microbiol. 1984;30(6):746-752
65. Murry MA, Fontaine MS, Torrey JG. Growth kinetics and nitrogenase induction in Frankia sp. HFPArI 3 grown in batch culture. Plant Soil. 1984;78:61-78
66. Schwencke J. Rapid, exponential growth and increased biomass yield of some Frankia strains in buffered and stirred mineral medium (BAP) with phosphatidyl choline. In: Nitrogen Fixation: Proceedings of the Fifth International Symposium on Nitrogen Fixation with Non-Legumes, Florence, Italy, 10-14 September 1990. Springer Netherlands. 1991:615-619
67. Burggraaf AJP, Shipton WA. Studies on the growth of Frankia isolates in relation to infectivity and nitrogen fixation (acetylene reduction). Can J Bot. 1983;61(11):2774-2782
68. Newcomb W, Peterson RL, Callaham D, Torrey JG. Structure and host-actinomycete interactions in developing root nodules of Comptonia peregrina. Can J Bot. 1978;56(5):502-531
69. Mastronunzio JE, Huang Y, Benson DR. Diminished exoproteome of Frankia spp. in culture and symbiosis. Appl Environ Microbiol. 2009;75(21):6721-6728
70. Benoist PATRICK, Müller A, Diem HG, Schwencke J. High-molecular-mass multicatalytic proteinase complexes produced by the nitrogen-fixing actinomycete Frankia strain BR. J Bacteriol. 1992;174(5):1495-1504
71. Cheggour A, Fanuel L, Duez C, Joris B, Bouillenne F, Devreese B. et al. The dppA gene of Bacillus subtilis encodes a new d-aminopeptidase. Mol Microbiol. 2000;38(3):504-513
72. van Bergeijk DA, Terlouw BR, Medema MH, van Wezel GP. Ecology and genomics of Actinobacteria: new concepts for natural product discovery. Nat Rev Microbiol. 2020;18(10):546-558
73. Rigali S, Titgemeyer F, Barends S, Mulder S, Thomae AW, Hopwood DA, Van Wezel GP. Feast or famine: the global regulator DasR links nutrient stress to antibiotic production by Streptomyces. EMBO Rep. 2008;9(7):670-675
74. Planas A. Peptidoglycan Deacetylases in Bacterial Cell Wall Remodeling and Pathogenesis. Curr Med Chem. 2022;29(7):1293-1312
75. Aspell T, Khemlani AHJ, Tsai CJY, Loh JMS, Proft T. The Cell Wall Deacetylases Spy1094 and Spy1370 Contribute to Streptococcus pyogenes Virulence. Microorganisms. 2023;11(2):305
76. Geelen D, Leyman B, Mergaert P, Klarskov K, Van Montagu M, Geremia R, Holsters M. NodS is an S-adenosyl-l-methionine-dependent methyltransferase that methylates chitooligosaccharides deacetylated at the non-reducing end. Mol Microbiol. 1995;17(2):387-397
77. Grohmann E, Christie PJ, Waksman G, Backert S. Type IV secretion in Gram-negative and Gram-positive bacteria. Mol Microbiol. 2018;107(4):455-471
78. Costa TR, Harb L, Khara P, Zeng L, Hu B, Christie PJ. Type IV secretion systems: advances in structure, function, and activation. Mol Microbiol. 2021;115(3):436-452
79. Hubber A, Vergunst AC, Sullivan JT, Hooykaas PJ, Ronson CW. Symbiotic phenotypes and translocated effector proteins of the Mesorhizobium loti strain R7A VirB/D4 type IV secretion system. Mol Microbiol. 2004;54(2):561-574
80. Sugawara M, Epstein B, Badgley BD, Unno T, Xu L, Reese J, Sadowsky MJ. Comparative genomics of the core and accessory genomes of 48 Sinorhizobium strains comprising five genospecies. Genome Biol. 2013;14:1-20
81. Gómez-Gómez L, Boller T. FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol Cell. 2000;5(6):1003-1011
82. Silipo A, Molinaro A, Sturiale L, Dow JM, Erbs G, Lanzetta R. et al. The Elicitation of Plant Innate Immunity by Lipooligosaccharide of Xanthomonas campestris. J Biol Chem. 2005;280(39):33660-33668
83. Zipfel C, Kunze G, Chinchilla D, Caniard A, Jones JD, Boller T, Felix G. Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell. 2006;125(4):749-760
84. Meneses N, Taboada H, Dunn MF, Vargas MDC, Buchs N, Heller M, Encarnación S. The naringenin-induced exoproteome of Rhizobium etli CE3. Arch Microbiol. 2017;199:737-755
85. Aslam SN, Erbs G, Morrissey KL, NEWMAN MA, Chinchilla D, Boller T. et al. Microbe-associated molecular pattern (MAMP) signatures, synergy, size and charge: influences on perception or mobility and host defence responses. Mol Plant Pathol. 2009;10(3):375-387
86. Soto MJ, Domínguez-Ferreras A, Pérez-Mendoza D, Sanjuán J, Olivares J. Mutualism versus pathogenesis: the give-and-take in plant-bacteria interactions. Cell Microbiol. 2009;11(3):381-388
87. Chabaud M, Gherbi H, Pirolles E, Vaissayre V, Fournier J, Moukouanga D. et al. Chitinase-resistant hydrophilic symbiotic factors secreted by Frankia activate both Ca2+ spiking and NIN gene expression in the actinorhizal plant Casuarina glauca. New Phytol. 2016;209(1):86-93
Author contact
![]() Corresponding author: Maher Gtari, E-mail: maher.gtarirnu.tn; Phone: +216 71 70 38 29; Fax: 216 71 70 43 29.
Corresponding author: Maher Gtari, E-mail: maher.gtarirnu.tn; Phone: +216 71 70 38 29; Fax: 216 71 70 43 29.