Journal of Genomics
ISSN: 1839-9940
ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2017; 5:36-47. doi:10.7150/jgen.16685 This volume Cite
Research Paper
Metagenomic Analysis of a Complex Community Present in Pond Sediment
Vivek Negi, Rup Lal ![]()
Molecular Biology Lab, Department of Zoology, University of Delhi, Delhi-07, India
Published 2017-3-10
Citation:
Negi V, Lal R. Metagenomic Analysis of a Complex Community Present in Pond Sediment. J Genomics 2017; 5:36-47. doi:10.7150/jgen.16685. https://www.jgenomics.com/v05p0036.htm
Other stylesAbstract

The metagenomic profiling of complex communities is gaining immense interest across the scientific community. A complex community present in the pond sediment of a water body located close to a hexachlorocyclohexane (HCH) production site of the Indian Pesticide Limited (IPL) (Chinhat, Lucknow) was selected in an attempt to identify and analyze the unique microbial diversity and functional profile of the site. In this study, we supplement the metagenomic study of pond sediment with a variety of binning approaches along with an in depth functional analysis. Our results improve the understanding of ecology, in terms of community dynamics. The findings are crucial with respect to the mechanisms such as those involving the lin group of genes that are known to be implicated in the HCH degradation pathway or the Type VI secretory system (T6SS) and its effector molecules. Metagenomic studies using the comparative genomics approach involving the isolates from adjacent HCH contaminated soils have contributed significantly towards improving our understanding of unexplored concepts, while simultaneously uncovering the novel mechanisms of microbial ecology.
Keywords: Functional diversity, pond sediment, genomic variation.
Introduction
Environmental pollution caused by the presence of highly persistent organic pollutants (POPs) has been identified as a problem at several places in Lucknow, India (7, 35). The presence of pesticides, especially those of organochlorine (OC) and organophosphate (OP) origin, in the soil significantly impacts our ecosystem. Although these pesticides might be present in low concentrations but due to biomagnifications they acquire the potential to introduce mutagenic and/or carcinogenic changes. Soil is naturally rich for the several uncultured microbes that aid, directly or indirectly, in the metabolism of these pesticides and their conversion to safer compounds. From this point of view, the field of metagenomics has been instrumental in elucidating and characterizing microflora belonging to different environmental niches. The studies have revealed the diverse functional contribution of the microbial community towards the environment. These contributions and functional roles include acid mine drainage [1], presence of bacterial rhodopsin in the marine metagenome [2], and the existence of degradation pathways for xenobiotic compounds and other unique cellular processes as required in stressed niches [3, 4, 5].
The extensive analysis of the cultivable [6, 7, 8, 9, 10, 11, 12] and uncultivable diversity [4, 5] of the HCH dumpsite located at the Ummari village in Lucknow, India, has revealed the presence of several potent HCH degraders as well as several HCH tolerant bacterial species. lin Genes, a group of genes involved in catabolic activities, are known to mediate the degradation of HCH isomers [13, 14, 15, 16] as well as play a major role in the degradation of xenobiotic compounds [17]. A comparative metagenomic survey of HCH contaminated soil was carried out considering different gradients, such as DS (dumpsite) (450 mg/kg of ∑HCH), 1 Km (0.7 mg/kg of ∑HCH) and 5 Km (0.03 mg/kg of ∑HCH), to determine the microbial dynamics active at stressed site. The comparative metagenomics presents new opportunities to access significant difference and similarity between communities. It also highlights the importance of functional identity for communities existing in specific environments. At such sites, the selection pressure of xenobiotic compounds is linearly correlated to increase in genome size and acquisition of foreign genes. Thus the environment specific and strain specific traits in these genomes have been well implicated in ancestral evolution and positive selection of bioremediation traits [4, 5].
Pond sediment has been previously reported to harbor pesticides such as lindane (57.6 ng/mL), α-endosulfan (80.9 ng/mL), β-endosulfan (220.9 ng/mL), chlorpyriphos (20.9 ng/mL), monocrotophos (8.3 ng/mL), dimethoate (2.9 ng/mL) and malathion (1.6 ng/mL) (35). As per the physiochemical analysis conducted by our group, the HCH content of pond sediment was found to be very low (> 0.909 mg/kg). In this study, we performed a metagenomic analysis of pond sediment (Chinhat village, Lucknow, India) with comparative genomics of isolates from the hexachlorocyclohexane (HCH) dumpsite (Ummari village, Lucknow India) or the pond sediment. In order to decipher the unique mechanisms present within the complex community, we investigated the community structure as well as the genetic structure in the context of the environment. Thus, in an attempt to elucidate the role of microbes in balancing the soil ecosystem, especially in contaminated sites, we (i) performed differential taxonomic binning of the pond sediment metagenome; (ii) compared the functional potential of pond sediment metagenome with that of HCH gradient metagenomes; (iii) conducted a metagenomic reassignment of reads with the help of genomic isolates from the same site or the pond sediment; and (iv) constructed a profile of genomic variation of the pond sediment based on essential single copy genes.
Materials and Methods
Sample Collection and Downstream Physiochemical Analysis
Two sediment samples were collected in triplicate from a pond located near the Chinhat village in Lucknow, India (26° 54ʹ 34.86 ̋ N, 81° 04ʹ 25.22 ̋ E), which is a few meters away from the IPL (India Pesticides Ltd.) factory. Collection was done in the month of January 2011. Weather conditions prevalent at the time of collection were as follows: 20 °C temperature, 0% precipitation, 38% humidity, 15 km/h wind and 966.24 mm average annual rainfall [Lucknow Centre, Geological Survey of India (GSI) Training Institute, Aliganj, Lucknow, India]. The samples were kept in sterile plastic bags, sealed and transported on ice (4°C) and thereafter stored at -80 °C until processed. The physiochemical properties of the sediment samples were analyzed at the Soil Testing Services - Indian Agricultural Research Institute. The extraction and analysis of HCH residues from samples were conducted as per the method described previously by Concha-Grana et al. A recovery of 74.95% with standard deviation of ±1.5% was obtained.
Metagenomic DNA Extraction and Sequencing
Power Max® Soil DNA Isolation Kit (MO-BIO, Carlsbad, CA, USA) was used for the isolation of DNA from the pond sediment samples in triplicate. Equal concentrations from each sample were pooled separately for two PS samples to reveal wide variety and range of the microbial community present in the pond sediment. DNA purity and concentration (PS1 = 1200 ng μL-1, PS2 = 1178 ng μL-1) were estimated using a NanoDrop Spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA). The two samples were then proceeded for DNA sequencing.
Clean sequencing data were obtained using the Illumina HiSeq 2000 platform (~28,164,307 and ~28,044,108 paired-end reads of PS1 and PS2, respectively). The data were generated by the removal of adaptors and reads containing the 'N' annotation from the raw data. The insert length and average read length of clean data were 170 bp and 91 bp, respectively.
De-novo Assembly of Pond Sediment Metagenome
The sequence data (Illumina HiSeq 2000 clean data) were assembled using the Velvet_1.2.03 [19] software at an optimized k-mer length of 39 (insert length, 170 bp; expected coverage, auto; minimum contig standard deviation for insert length, 20 bp; and length cutoff, 200 bp). The k-mer length was optimized after running Velvet at different k-mer lengths. The CAST algorithm was used to filter the assembled sequences [20]. The cutoff for minimum contig length was kept >200 bp in order to ensure a reasonable length for predicted ORFs [3].
Comparative Taxonomic Binning of Metagenomes
Assembled contigs were binned using RAIphy [21] and also by comparing the same to the NCBI non-redundant protein database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr, June, 2015) using BLASTX (E-value = 1e-10) [22]. The MEtaGenome ANalyzer (MEGAN 5.0) [23] was used at default parameters in order to assign contigs to phylogenetic groups at the genera level. Six metagenomes were classified (PS1, PS2, 1 Km, 5 Km, DS(Dumpsite) and SolexaDS) against the nr database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr, June, 2015) (BLASTX, E-value = 1e-10) with and without the isolates (Table S4) obtained from the HCH contaminated site or from other suitable sites of pond sediment. Differential abundance of bacterial taxa at the phylum level among PS1 and PS2 samples were statistically computed using METASTATS (http://metastats.cbcb.umd.edu). The total numbers of subjects were two with 0.05 threshold p value and 1000 permutations. For each group, mean values for the present phyla were computed along with variance and standard error.
Genomic Variation in Pond Sediment Metagenome
For the analysis of the genomic variation present in pond sediment, 74 of the most abundant prokaryotic genomes obtained from taxonomic binning of pond sediment samples and whole genomes isolated from the HCH dumpsite were shortlisted as the set of reference genomes (Table S4). A set of 107 essential single-copy gene HMMs (24) were searched against the predicted ORFs of pond sediment associated genomes using HMMER3 (http://hmmer.janelia.org/). Out of these 107 genes, a set of 60 essential single-copy genes were found to be in consensus with all selected genomes (Table S5). These marker genes, which were deemed capable of resolving global phylogeny, were then used to determine genomic variation in pond sediment with respect to the reference genome marker genes. All associated analytical procedures were performed using R v. 3.2.2 via the ace function of ape. Principle component analysis of tetranucleotide frequencies of the marker genes was conducted on the genomes and the pond sediment sample in order to form clusters. To identify genomic variation within pond sediment samples, we used a combination of principle components that display the clusters in question. To estimate and analyze the pattern of genotypic diversity, we also performed phylogenetic principal components analysis (PCA) using a maximum-likelihood algorithm for essential single copy genes. The variation in genotypic diversity present within the pond sediment is a function of the complexity of the community. The plot was constructed along two pairs of axes; it was composed of all terminal and internal phylogenetic nodes with the branches connecting the adjacent nodes. The R plot visualization also includes the branches and nodes connecting clades.
Gene Prediction and Functional Annotation
MetaGene gene finder [26] and the metagenome version of MetaProdigal [27] were used to predict open reading frames (ORFs) of the metagenomic contigs. The functional annotation of these predicted ORFs was carried out by database comparison searches. HMMER3 was used to assign Reverse Position-Specific BLAST (RPS-BLAST) of peptide sequences present in pond sediment metagenomes to COG clusters [28] and PFAM families [29] present in the NCBI Conserved Domain Database (E-value < 1e-5 and Percentage Identity > 20%). Pathway annotation of peptide sequences was done against the KEGG GENES [30] database using Reverse Position-Specific BLAST (RPS-BLAST) [22] (Bit Score > 60 and bi-directional hit rate (BHR) > 0.95). Rarefaction curve, two sided Fisher's exact test and Storey's FDR methods were carried out on the Pfam [29] database using STAMP [31]. Principle Component Analysis (PCA) of COG categories of the metagenomic samples was conducted using R v. 3.2.2 [https://cran.r-project.org/bin/windows/base/old/3.2.2/]. Shannon diversity for each metagenomic sample was calculated for functional COG categories and microbial diversity by EGT analysis in R using the vegan [32] package. MinPath [33] was used for predicting probable pathways. All-versus-all BLASTP (default parameters) followed by MCL clustering (http://micans.org/mcl/) was used to determine clusters of homologs in order to validate the pathways.
Data Availability
The data obtained post-analysis of the pond sediment was submitted to NCBI SRA under the accession ID SRP047092. Metagenomic sequence data obtained from previous metagenomic reads have been deposited at MG-RAST ID under the study accessions: DS =4461840.3, 1 Km =4461013.3 and 5 Km =4461011.3 and at GenBank/DDBJ/EMBL : SolexaDS = ERP001726.
Results
Sequence data generation and assembly
The metagenomic data obtained after analyzing the pond sediment from Chinhat, Lucknow resulted in 106 × 106 Illumina HiSeq 2000 paired-end reads of PS1 and PS2 each. These paired-end reads were arrived at after quality filtering raw data for adapter sequences and 'N' nucleotide reads. After assembling the clean data on Velvet_1.2.03 [19] at an optimized k-mer length of 39,774,416 and 1,483,949 contigs with n50; 158 and 116 of PS1 and PS2 samples were obtained, respectively (Table 1). The percentage of reads used for the assembly process was 50.17% for PS1 and 30.25% for PS2. The quality of assembly was checked by CAST algorithm to obtain good quality of sequence for the analysis of complex communities present in the metagenomic samples. In total 243,624 and 206,869 ORFs were predicted for PS1 and PS2, respectively.
Table 1
Sequencing Data and sequence assembly statistics of both pond sediment samples.
| Sample | PS1 | PS2 |
|---|---|---|
| Total no. of reads | 106,039,716 | 175,086,320 |
| Percentage of reads used | 50.17 % | 30.25 % |
| Total Assembly length (bp) | 59,521,231 | 51,183,827 |
| K-mer | 39 | 39 |
| No of Contigs obtained | 774,416 | 1,483,949 |
| n50 | 158 | 116 |
| Maximum length of Contig (bp) | 10,214 | 10,340 |
| GC Content of Contigs | 62 ±8 % | ±9 % |
Physiochemical Properties of the Sediment
The electrical conductivity across the pond sediment [1.10 dS/m (PS1) and 0.73 dS/m (PS2)] was lower as compared that obtained for DS (8.5 dS/m) [4]. Along similar lines, when compared to DS, the potassium content of the pond sediment was <10% while the salinity was within the normal range (PS1 = 89 kg/ha and PS2 = 90 kg/ha; DS: 918 kg/ha) [4]. Physiological properties of the pond sediments (PS1 and PS2) exhibited significant differences (p<0.00001) based upon the different HCH contaminated locations (DS, 1 Km and 5 Km) (Table 2) [4]. The residue of HCH isomer contamination detected in the sediment samples is elucidated in Table 2. The collective concentration of all HCH isomers found in the pond sediment was 0.905 ±0.003 mg/kg (PS1) and 0.909 ±0.006 mg/kg (PS2). In all the sediment samples collected for this study, it was found that the β-HCH and α-HCH isomer content was less than 0833 ±0.003 mg/kg and greater than 0.076 ±0.002 mg/kg, respectively.
Table 2
Chemical properties and HCH Residue Analysis (mg/kg) of soil samples. pH values of the samples are neutral and normal salinity levels are represented by low concentration of cations (except Nitrogen) and Electrical Conductivity.
| Samples | PS1 | PS2 |
|---|---|---|
| pH | 7.72 | 6.92 |
| Electrical Conductivity (dS/m) | 1.10 | 0.73 |
| Organic Carbon (%) | 0.85 | 0.41 |
| Available Phosphorus (kg/ha) | 25.3 | 24.9 |
| Available Potassium (kg/ha) | 89 | 90 |
| Available Nitrogen (kg/ha) | 439 | 502 |
| Salinity | Normal | Normal |
| αHCH (mg/kg) | 0.074 | 0.076 |
| βHCH (mg/kg) | 0.831 | 0.833 |
| ΣHCH (mg/kg) | 0.905 | 0.909 |
Phylogenetic Reassignment of Metagenomic Reads using Sequenced Whole Genomes from Contaminated Sites
The percentage of assigned reads was higher when the metagenomic reads were aligned to a nonredundant (nr) database along with the whole genome data rather than a nonredundant (nr) database only (p value < 0.0001) (Fig. 1a). In pond sediment metagenome, the improved metagenomic reassignment was computed to be 1.02%, whereas the same cumulatively for all the metagenomes was determined to be 0.44%. Percentage of reads reassigned to major phyla in pond sediment were as follows: Proteobacteria [16.35%], Actinobacteria [7.67%], Chloroflexi [5.48%], Acidobacteria [4.45%], Planctomycetes [4.22%], Verrucomicrobia [2.48%], Firmicutes [2.38%], Cyanobacteria [1.79%], Thaumarchaeota [0.92%], Euryarchaeota [0.35%], Deinococcus-Thermus [0.28%], Nitrospirae [0.29%], Armatimonadetes [0.25%], Spirochaetes [0.27%], Candidatus [0.12%], Chlorobi [0.06%], Poribacteria [0.11%], Aminicenantes [0.08%] and Ignavibacteriae [0.12%] (Table S1). Most of the bacteria belong to phylum Proteobacteria, and whole genome sequence information is available for maximum members of this phylum, therefore the outcome of the analysis for this group is best. In addition, this methodology also highlights comparatively rarer phyla such as Candidatus, Cyanobacteria, etc.
Pond Sediment Microbial Diversity Analysis
Phylum level of taxonomic assignment as obtained from BLASTP was displayed using MEGAN (E-value ≥ 1e-5) (Fig. S1). The cumulative number of unique NCBI non-redundant protein (y-axis) was plotted as the function of the cumulative number of percentage of contigs (x-axis). The curve for PS1 and PS2 becomes flat at the right side, which implies that the abundance of the non-redundant protein family had reached a saturation level (Fig. S2). Additionally, by using ggplo2 in R v. 3.2.2, a stacked area plot was constructed in order to show that pond sediment has an abundance of microbes spread over a range of phyla. All the components were stacked in this plot (Fig. 1b). p-Value and q-value were chosen in the top order to resolve individual patterns. The relative abundance of dominant bacterial phyla in the pond sediment samples are represented in the plot along with median values, variance, standard error, p-value and q-value. (Table S2) The majority of the microbiota present in pond sediment includes the degraders and contaminant resistant and antibiotic producing microbes. Taxonomical classification across PS1 and PS2 was determined using the Environmental Gene Tag analysis (EGT: a compositional based binning) as well as BLASTP of contigs against NCBI's non-redundant protein database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr, June, 2015). According to EGT analysis, the PS1 and PS2 samples have similar profiles (R2 = 0.99). Furthermore, as EGT typing assay annotates the abundance of microbes by presenting an average percentage for each genera along with standard deviation, the following bacteria were identified as the most abundant in both PS1 and PS2 (R2 > 0.99): Corynebacterium (4.33% ±0.53), Rhodococcus (3.06% ±0.90), Bradyrhizobium (2.62% ±0.84), Sorangium (2.25% ±0.99), Thauera (0.94% ±1.41), Methylibium (1.02% ±1.54), Candidatus (2.24% ±3.40), Anaeromyxobacter (5.83% ±2.73), Streptomyces (5.82% ±4.46) and Burkholderia (12.61% ±3.56) (Table S3).
Fig 1
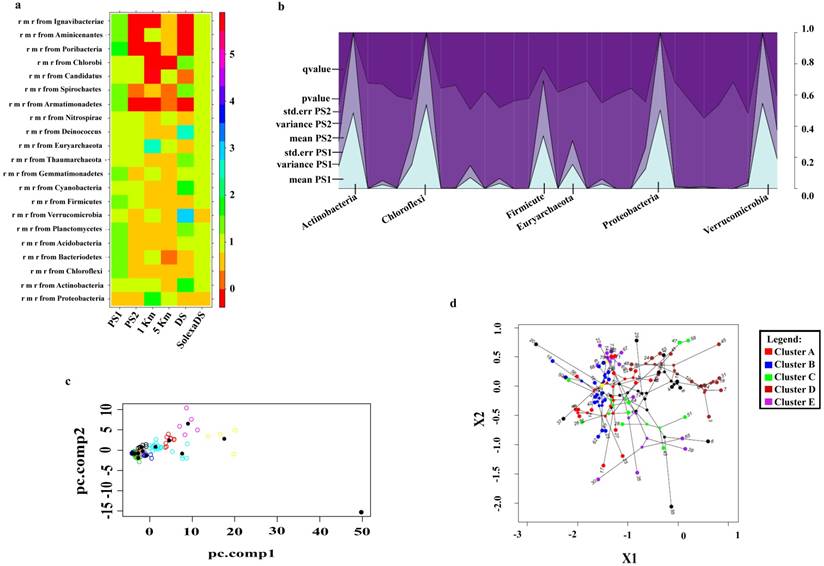
Differential taxonomic binning of pond sediment. a) Phylogenetic reassignment of metagenomic reads of PS1, PS2 and HCH gradient (1 Km, 5 Km, DS and SolexaDS) against the nr-database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr, June, 2015) (BLASTX, E-value = 1e-10), b) Stack area plot of the most abundant phyla present among PS1 and PS2 samples which were statistically computed using METASTATS (P value ≤ 0.05 and 1000 permutations), c) Depiction of genomic variation in pond sediment by principal component analysis of tetranucleotide frequency of essential single copy genes using R v. 3.2.2 via the ace function of ape. pc.comp1 and pc.comp2 are its two principle components. d) The plot was constructed along two principle components; X1 and X2 were composed of all terminal and internal phylogenetic nodes with the branches connecting adjacent nodes. X1 and X2 axis in the principle component plot are the eigenvectors with the highest eigenvalues for essential single copy genes of the most abundant bacterial phyla. Clustering of nodes represented the genomic variation in pond sediment and Cluster B was found to be abundant in pond sediment metagenome.
Genomic Variation within the Pond Sediment Metagenome
The most abundant genomes present within the complex pond sediment communities (Table S4) were identified by clustering 60 essential single copy genes (Table S5) present in the two principle components. The tetranucleotide frequency of essential single copy gene based clusters, as generated by principle component analysis, has proved to be immensely useful in identifying genomes from the metagenome of sediments. A large majority was found to be composed of organisms that are actively evolving to compete and proliferate in synchrony with the microenvironment of the soil (Fig. 1c). However, despite the several tools used for the purpose, many of the genomes present in the clusters could not be clearly demarcated. In order to distinguish between the genotypic diversity present within the pond sediment, we used a combination of principle components by utilizing a maximum-likelihood algorithm that gave us the greatest possible resolution between the genomes (Fig. 1d). In this study, we have demonstrated that genotypic diversity and clustering of genomes can be used to predict the magnitude by which each genome is related to the other and can also be used to reveal how one cluster achieved greater diversity than the other. The differential pattern of clusters and the position of the internal nodes are predictive of the mean distance between adjacent nodes; these values are known to be relatively different within the clusters. The ability of a null Brownian model of genotypic diversity to generate clusters with the observed difference in mean length between the nodes can be tested via simulation. The clusters that are being compared have either diversified under differing sequence of single copy essential genes or remain conformed; this is checked for by the rejection of the simulated Brownian null model for genotypic diversity for the pond sediment. The clusters that are packed more densely in the plot show lower genotypic diversity as compared to the clusters with diffused nodes. Clusters A, C and D appear to have continuously expanded to a larger number of regions whereas clusters B and D are confined only to one region. Each cluster is denoted with a different color and the pond sediment appears to be well associated with cluster B. The Brownian rate parameter was higher in clusters A, C and E than in clusters B and D.
Comparative Functional Annotation of Contaminated Sites
In an attempt to delineate the interaction of the bacteria present in pond sediment [7, 34, 35, 36] with their environment as well as among themselves, we explored their metabolic potential using KEGG (Kyoto Encyclopedia of Genes and Genomes, version 69) [30], PFAM (Pfam 27.0, Protein families database) [29] and COG (Cluster of Orthologs Groups) [28] (Fig. 2). In line with previously published metagenomic studies [4, 5], pond sediment metagenome was also found to contain a variety of subsystem proteins coding for membrane transport, cell signaling, chemotaxis, transposases, and phage elements (Table S6). Pond sediment metagenome also contained genes that code for proteins of plasmid related function and resistance against toxins as well as antibiotics. The KEGG analysis results were used to map enzymes present in pond sediment metagenome into the various metabolic pathways that they were involved in. These pathways included the bacterial secretory system, the aromatic compound metabolism (degradation of chlorocyclohexane, chlorobenzene, toluene, catechol, terephthalate and xylene isomers) system and the two component system. Principal component analysis (PCA) was performed on the COG categories of all metagenomic samples (PS1, PS2, DS, 1 Km, 5 Km and SolexaDS) (4, 5) (Fig. 2c). To compare the variability present among the metagenomic samples, we separately calculated the coefficient of variation (CV) within each sample for both functional as well as EGT analysis. Finally, Shannon diversity was calculated between samples for computing functional (COG analysis) and taxonomic diversity (EGT analysis). Solexa sequencing of the dump site (SolexaDS) revealed the presence of a high degree of diversity in microbes as well as in the functional subsystems of the community (Fig. 2d). We found that pond sediment is less diverse than the three HCH gradients.
According to the gene centric approach for sequencing of the contaminated sites, several enzymes involved in the degradation of chlorocyclohexane and chlorobenzene (ko00361) were found to be present in the pond sediment metagenome. These enzymes mostly belong to the families of hydrolases, oxidoreductase, isomerases, and lyases (Table S7), which assist microbes in tolerating and metabolizing xenobiotic compounds. Genes implicated in HCH (Hexachlorocyclohexane) degradation, especially those involved in the upper pathway, are present in pond sediment metagenomes that are enriched with sequences for the linA, linB (Evalue > 1e-5) genes (Evalue > 1e-5).
Genes involved in T6SS were abundant in the pond sediment metagenome and were assigned to the KEGG Orthology (KO) system [30] using reverse position-specific (RPS) BLAST [22] against the KEGG GENES database with a BLAST score (bit score) greater than 60 and a bi-directional hit rate (BHR) of > 0.95 (Fig. S4) (Table S8).
Discussion
Physiochemical Properties of the Pond Sediment
The presence of a relatively lower electrical conductivity level, salinity, and ion concentration of PS1 and PS2 sediment samples indicated towards positive microbial activity and a reduced stress microbial community that operated with a high metabolic efficiency rate (Table 1). The physiochemical conditions of the pond sediment match well with the condition of bacteria (predicted by EGT analysis) (Fig. 1) derived from the site of isolation [35]. As per the Stockholm Convention of 2009, α-HCH, β-HCH and γ-HCH are considered as persistent organic pollutants. Residue levels of HCH isomers and total HCH obtained from different pond sediment sites were computed during the course of this study; the results indicated towards low levels of HCH contamination (Table 2). The presence of HCH contamination in the pond sediment near the Indian Pesticide Limited (IPL) factory at Chinhat, Lucknow is concordant with the fact that the factory discharges its waste (HCH muck) into the IPL drainage system, which later enters the pond. This phenomenon results in distribution of the contaminant in the pond water as well as in the bank (sediment). The waste produced at the plant comprises predominantly of α- and β-HCH residues. The lindane production plant produces waste that can be classified under two categories: (a) bulk waste fraction comprising mainly of α- and β-HCH isomers; and (b) minor waste fraction, which contains γ- and δ-HCH isomers [37]. The prevalence of HCH was less than the permissible limit (>4000 mg/kg for t-HCH). However, the recalcitrant nature and stability of β-HCH and δ-HCH in sediment can lead to its biomagnification and harm to nature.
Fig 2
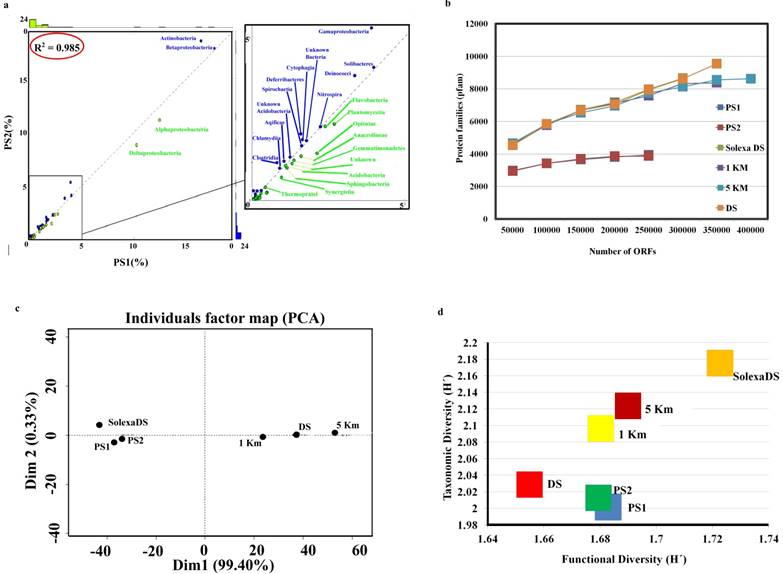
Functional Annotation from pond sediment and sites across dumpsite. a) Extrapolation of taxonomic diversity of PS1 and PS2 (Fisher's Exact Test, P > 0.01) is the overall representation of phylum profiles using Pfam protein domains and the associated Gene Ontology (GO) in PS1 versus PS2 (R2 = 0.985), b) Rare fraction analysis performed on the unique Pfam protein domains across pond sediment (PS1 and PS2) and three HCH gradients (1 Km, 5 Km, DS(Dumpsite) and SolexaDS), c) Principle Component Analysis (PCA) plot of NCBI COG categories of three HCH gradients and two samples of pond sediment metagenome to validate the functional profiling of matagenomic samples, d) Plot showing Shannon diversity across pond sediment (PS1 & PS2), 1 Km, 5 Km, DS(Dumpsite) and SolexaDS for functional and diversity (by EGT) analysis.
Differential Binning of Pond Sediment
Phylogenetic origins of a majority of metagenomic reads remain anonymous even after applying most of the popular and credited binning algorithms. The enormous size of the metagenomic sample is one of the crucial factors that interfere with the classification of its reads largely due to the biased similarity with reference genomes in databases. In order to improve the efficiency of the metagenomic binning process, we introduced the concept of metagenomic reassignment of reads. The first step was to annotate the six metagenomes (PS1, PS2, 1 Km, 5 Km, DS and SolexaDS) against nr-database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr, June, 2015). Then the annotation was performed against the nr-database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr, June, 2015) with the isolates (Table S4) that were obtained from the HCH contaminated site or the pond sediment. The metagenomic reads that were phylogenetically reassigned during the second step were determined and reported in percentage. In pond sediment metagenome, the improved metagenomic reassignment was computed to be 1.02%, whereas the same for all the metagenomes was cumulatively determined to be 0.44%. The use of the refined binning methodology on the pond sediment samples gave the following results: cellular organisms- 61.43%, unassigned reads- 1.98% and, no hit- 2%. It was determined that the cellular organism component in pond sediment predominantly included bacteria [56.39 %], archaea [0.69 %] and eukarya [0.86 %]; it is evident that the presence of bacteria-based communities was in dominant majority (Fig. S3).
The dominant bacterial phyla in pond sediment were identified and these, along with other unknown phyla, were subjected to METASTATS (http://metastats.cbcb.umd.edu) in order to compute differential abundance. The most significant (P-value ≤ 0.05) as well as abundant phyla were explored as a part of this study. Statistically significant phyla that were displayed in stacked plot of R v. 3.2.2 were as follows: Proteobacteria (P-value = 7.8 × 10-7, Q-value = 2.7 × 10-6), Actinobacteria (P-value = 1.0 × 10-26, Q-value = 7.0 × 10-26), Euryarchaeota (P-value = 8.4 × 10-3, Q-value = 2.3 × 10-2), Chloroflexi (P-value = 4.1 × 10-17, Q-value = 1.9 × 10-16), Verrucomicrobia (P-value = 1.7 × 10-2, Q-value = 3.3 × 10-2), Planctomycetes (P-value = 3.1 × 10-2, Q-value = 4.7 × 10-2), Chlorobi (P-value = 1.9 × 10-2, Q-value = 3.3 × 10-2) and Acidobacteria (P-value = 1.7 × 10-2, Q-value = 3.2 × 10-2). All of the above mentioned phyla, especially the Proteobacteria, were predominantly found in the soil fraction. Taxonomical distributions, such as the one outlined above, are in context of the environment in question. Community structure of pond sediment is known to be extremely complex and only a few dominant phyla are capable of thriving under such severely competitive environmental conditions.
In order to determine which genera were the most active and abundant in the pond sediment, genomic variation was analyzed using essential single copy genes [24]. These 107 essential single copy genes are known to be highly conserved; examples of the same include the ribosomal genes or the conserved core genes. The top 74 genera that were selected from the pond sediment were found to be most abundant as well as to contain all 60 such essential single copy genes (Table S4). It was observed that the pond sediment was close to cluster B and that the genera dominating this cluster were Beutenbergia, Brachybacterium, Cellulomonas, Cellvibrio, Clavibacter, Isoptericola, Kineococcus, Kitasatospora, Kribbella, Kytococcus, Microbacterium, Micrococcus, Nocardioides, Sanguibacter, Streptomyces, Thermobispora and Xylanimonas (Fig. 1d). These genera are found abundantly in soil specimens and are possibly enriched in the pond sediment due to presence of aromatic hydrocarbons and some other similar factors. The functional properties of the pond sediment are in accord with the presence of genes involved in aromatic compound degradation, secretion, and transport.
Statistical Analysis
Various statistical analyses were performed to prove and validate the significance of metagenomic samples with complex communities. In terms of the Pfam protein domains, the functional profiles of samples PS1 and PS2 are closely correlated to each other (R2 = 0.985) (Fig. 2a). The results also highlight the abundance of bacteria belonging to the phylum Actinobacteria and Betaproteobacteria in PS1. Actinobacteria consist of bacterial groups that span a wide array of morphology and are known to demonstrate highly variable physiological and metabolic profiles. Betaproteobacteria on the other hand have been previously reported to degrade xenobiotic compounds as well as to fix nitrogen. PS2 sample was found to be dominated by Alphaproteobacteria and Deltaproteobacteria. These phyla represent phototrophs, plant symbionts and are composed of predominantly of aerobic bacteria.
The solid lines with different colors in the rarefaction curve represent ≤ 4 × 106 ORFs each across Dump Site and pond metagenomes (Fig. 2b). The cumulative number of Pfam protein families (y-axis) is plotted as the function of the cumulative number of ORFs (x-axis). The curve for PS1, PS2, 1 Km and 5 Km becomes flat at right, which implies that the abundance of the Pfam protein family has reached a saturation level. However, in case of DS, the slope of the curve was steep, hence there is some scope to determine further the protein family richness. The demography of PS1, PS2, and SolexaDS forms one cluster (Fig. 2c). This can be attributed to similar sequencing data (Illumina Hiseq) and a significantly correlated cluster of ortholog (COG) profiles of respective samples (R2 > 0.75). To compare the variability in Shannon diversity of the metagenomic samples, coefficient of variation was calculated for functional (COG) as well as taxonomic diversity (Fig. 2d). The HCH gradients show a linear pattern, which is indicative of an increase in diversity as distance from DS increases. Pond sediment metagenomic samples show a higher functional diversity rather than taxonomic diversity. It might be due to the microbe-friendly conditions that are prevalent in the pond sediment, which lead to a competition wherein only the dominant genera survive. DS sequencing methodology can be held responsible for the large changes that are observed in diversity. In case of SolexaDS, a very high diversity is observed and this can be due to HCH contamination. As the HCH contaminated site is not favorable for microbes, the possibility of dominant genera affecting the diversity at such sites is expected to be less.
Metabolic Profile of Pond Sediment
Soil is the most complex community known till date and its diversity remains to be adequately decoded and understood. Even with the help of high throughput technologies (e.g., Illumina, Pyrosequencing and Sanger) and the presence of advanced soil DNA extraction kits (MoBio), delineating complete and comprehensive information regarding soil remain a tough challenge; however, it is possible to make some assumptions and generalizations regarding metabolic pathways present at the site.
In the specific case of pond sediment, the most abundant subsystem includes factors implicated with amino acids and its derivatives, carbohydrates, cell division and cell cycle, cell wall and capsule, clustering-based subsystems, cofactors, vitamins, prosthetic groups, pigments, DNA metabolism, dormancy and sporulation, fatty acids, lipids, and isoprenoids, iron acquisition and metabolism, membrane transport, metabolism of aromatic compounds and several other processes (Table S6). These are the populated subsystems, which were assembled from analysis of the different microbes present in pond sediment. Detailed annotation of pond sediment environment helps in creation, curation, population, and exchange of subsystems within the community. Further in-depth functional study of pond sediment revealed a high abundance of microbial cation/multidrug efflux pump system, response regulator (CheY-like receiver, AAA-type ATPase, and DNA-binding domains) and the serine/threonine protein kinase enzyme. The same subsystems were prevalent in other metagenomes (DS, 1 Km, 5 Km and SolexaDS) as well. These trends are constant in all the above mentioned soil and sediment metagenomes.
Efflux pumps play an important role in enabling multidrug resistance as they function as an energy-dependent active transport system for pumping out unwanted toxic substances. The structure of the pump complex in bacteria is such that the unwanted toxic substance is exported directly into the external medium rather than into the periplasm. The bacterial cells are benefited by this mechanism because once the toxic substance has been expelled into the external space it then has to pass outer membrane again in order to reenter the cell. These pumps function in conjunction with other outer membrane barriers in order to defend the bacterial cell in extreme niches. The microbial response regulator consists of an N-terminal CheY-like receiver domain and a C-terminal DNA-binding domain. These response regulators are involved in regulating the production of important virulence factors, bacteriocins, and extracellular polysaccharides.
Table S7 of the supplementary information enumerates the enzymes implicated in degradation of the aromatic compounds chlorocyclohexane and chlorobenzene as obtained from the annotation profile of the pond sediment. The two enzymes found in pond sediment metagenome, maleylacetate reductase (EC 1.3.1.32) and hydroxyquinol 1, 2-dioxygenase (EC 1.13.11.37), are capable of catalyzing the conversion of hydroxyquinol to 3-oxoadipate when they act together. Enzymes of the modified ortho cleavage pathway yield chlorocatechols after degrading chlorinated aromatic compounds [38]. Maleylacetate is converted to 3-oxoadipate by maleylacetate reductase in an enzymatic pathway that is common to the metabolism of several aromatic compounds. A rich diversity is observed in case of dehalogenases owing to the ability of microorganisms to degrade a wide variety of halogenated compounds to different degrees. Focusing on the lin group of genes (Table S7) that are implicated in the degradation pathway of HCH isomers, it is well known that these genes diverge in order to catalyze several catabolic functions [39]. For example, linA encodes for HCH dehydrochlorinase and linB, belonging to α/β hydrolase family, encodes for halidohydrolase, which acts as the primary enzyme of the pathway. LinB has a very broad substrate preference for halogenated compounds, whereas the substrate range for LinA is restricted to α-, γ- and δ-HCH and their corresponding PCCH products [14]. linC encodes for a 2, 5-DDOL dehydrogenase and has an activity profile similar to that of the linX gene [40].
In nature, microbes thrive in complex communities where they interact with other species living within the same environment. The abundance of the type VI secretion system (T6SS) in pond sediment indicates towards a complex interaction between microbes and their environment with respect to transport mechanisms [41] (Fig. S4). This secretion system is a multicomponent complex that has a sec-independent mechanism for the transport of the effector proteins associated with it. The T6SS system translocates its effector proteins across recipient cells in a contact dependent manner [42, 42]. These well known and characterized effector proteins include enzymes that can act upon actin or degrade peptidoglycan. Many unknown effector molecules may be involved in different biotic interactions ranging from symbiosis to pathogenesis; this is an area that still needs to be researched and explored. T6SS is composed of 13 core subunits along with various additional components that are essential for the secretion process [44]; together these subunits and components form a cluster of genes that is approximately 20 kb size. According to results derived from functional analysis, protein localization experiments [45, 46] and bioinformatic analysis [44], the genes involved in this secretory system can be classified into three categories. The first category is composed of membrane associated proteins, integral membrane proteins and lipoproteins (K11892, K11891, K11906, K11894, K11910, K11911, K11893, K11907 and K11896). The second category (K11903, K11904, K11900, K11895) is composed of proteins that make up the tailed bacteriophage machinery that make up the cell puncturing device [47]. The last category (K11901) is composed of proteins whose function remains to be delineated. In addition to the abovementioned genes, several accessory genes (K11908, K11907, K02557, K11898, K01115, 03Y_06575) are also associated with the cluster of T6SS genes. It is noteworthy that within the T6SS machinery, genes comprising of the Virulence Associated Cluster (VAS) have both a regulatory as well as a structural significance [48].
An analysis of the relative functional subsystems present in pond sediment samples have highlighted some unique functional attributes (R2 = 0.985) that are specific to the system under study. Based on our results we can deduce that the complex community present within the pond sediment fosters an environment conducive to a high level of inter-specific competition. As a direct consequence of this, the microbes present in pond sediment have developed highly evolved subsystems for defense, survival, and resource gathering.
Conclusion
This study involved a physiochemical analysis of sediment, microbial diversity, and functional capability of the microbial community inhabiting pond sediment near IPL a well-known producer of lindane. The assembly of contigs plays an important role in diversity and functional analysis. Phylum, such as the Proteobacteria, gets assembled more conveniently as compared to Verrucomicrobia or Actinobacteria due the absence of regions that limit assembly. Thus, the information retrieved by high-throughput sequencing of complex communities might be biased towards certain phyla. Hence, in order to get a complete and comprehensive profile of the microbiota of complex communities, the comparative approach of metagenomics must be supplemented with genomic analysis. This study sheds light on the comparative functional profiling between DS, 1 KM, 5 KM and pond sediment in order to correlate the microbial community with its complexity. Besides HCH, there are many other aromatic compounds that also get discharged from the factory effluents into the soil, from where it leaches into the ground water thus contaminating the entire ecosystem. According to differential binning approaches Mycobacterium, Corynebacterium, Rhodococcus, Bradyrhizobium, Sorangium, Thauera, Methylibium, Candidatus, Anaeromyxobacter, Streptomyces and Burkholderia were the most abundant genera in the pond sediment samples collected by us. Most of the genera listed above are known to degrade aromatic compounds and have advanced secretory systems.
Distribution of T6SS in bacteria inhabiting environmental niches is not well documented. Metagenomic sequencing of the pond sediment dataset has helped us in understanding T6SS as well as the mechanism of export of effector molecules in these tightly regulated processes. Most of the elements present in pond sediment are involved in horizontal transfer of genetic material, owing to that, there is a very robust network for exchange of genetic information within the community. As evidence for the widespread exchange of genetic information in the pond sediment, it was seen that an analysis of the diversity of the pond sediment shows a high occurrence frequency for genera that harbor a large number of plasmids. Due to this, the microbes present at pond sediment have evolved a superior adaptability for the contaminated sites and are also actively involved in degradation of the toxic contaminants.
Until now, the studies have revealed the existence of very few pathways for microbial interaction but with increased research and improved knowledge along with the discovery of novel pathways, the concept of microbial interaction will reach a new level of understanding. However, understanding complex microbial communities such as those that exist in soil requires ultra deep sequencing (50 Tbp approx) for the extraction of useful information [49].
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The work was supported by grants from the Department of Biotechnology (DBT), Government of India, under project (BT/PR3301/BCE/08/875/11), the All India Network Project on Soil Biodiversity-Biofertilizers (ICAR) XIth Plan (12-A/2007-lA ll), DU-DST Promotion of University Research and Scientific Excellence (PURSE) grant and National Bureau of Agriculturally Important Microorganisms (NBAIM) NBAIM/AMASS 2014-17/PF/9/BG/378. We thank N.S., A.S., H.V. and N.G. for critically reviewing the manuscript. V.N. gratefully acknowledges the Council for Scientific and Industrial Research (CSIR) for providing research fellowship.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tyson GW, Chapman J, Hugenholtz P. et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37-43
2. Béjà O, Aravind L, Koonin EV. et al. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science. 2000;289:1902-1906
3. Albertsen M, Hugenholtz P, Skarshewski A. et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol. 2013;31:533-538
4. Sangwan N, Lata P, Dwivedi V. et al. Comparative metagenomic analysis of soil microbial communities across three hexachlorocyclohexane contamination levels. PLoS One. 2012;7:e46219
5. Sangwan N, Verma H, Kumar R. et al. Reconstructing an ancestral genotype of two hexachlorocyclohexane-degrading Sphingobium species using metagenomic sequence data. ISME J. 2014;8:398-408
6. Singh A, Lal R. Sphingobium ummariense sp. nov, a hexachlorocyclohexane (HCH)-degrading bacterium, isolated from HCH contaminated soil. Int J Syst Evol Microbiol. 2009;59:162-166
7. Dadhwal M, Jit S, Kumari H. et al. Sphingobium chinhatense sp. nov, a hexachlorocyclohexane (HCH) degrading bacterium isolated from an HCH dump site. Int J Syst Evol Microbiol. 2009;59:3140-3144
8. Bala K, Sharma P, Lal R. Sphingobium quisquiliarum sp. nov, P25T a hexachlorocyclohexane (HCH) degrading bacterium isolated from HCH contaminated soil. Int J Syst Evol Microbiol. 2010;60:429-433
9. Sharma P, Verma M, Bala K. et al. Sphingopyxis ummariensis sp. nov, isolated from hexachlorocyclohexane (HCH)-dumpsite in north India. Int J Syst Evol Microbiol. 2010;60:780-784
10. Kumar R, Dwivedi V, Negi V. et al. Draft Genome Sequence of Sphingobium lactosutens Strain DS20 isolated from a Hexachlorocyclohexane (HCH) Dumpsite. Genome Announc. 2013;1:e00753-13
11. Negi V, Lata P, Sangwan N. et al. Draft genome sequence of hexachlorohexane (HCH) degrading Sphingobium lucknowense strain F2T isolated from the HCH dumpsite. Genome Announc. 2014;2:e00788-14
12. Verma H, Kumar R, Oldach P. et al. Comparative genomic analysis of nine Sphingobium strains: insights into their evolution and hexachlorocyclohexane (HCH) degradation pathways. BMC Genomics. 2014;15:1014
13. Nagata Y, Endo R, Ito M. et al. Aerobic degradation of lindane (γ-hexachlorocyclohexane) in bacteria and its biochemical and molecular basis. Appl Environ Microbiol. 2007;76:741-752
14. Kumari R, Subudhi S, Suar M. et al. Cloning and characterization of lin genes responsible for the degradation of hexachlorocyclohexane isomers by Sphingomonas paucimobilis strain B90. Appl Environ Microbiol. 2002;68:6021-6028
15. Lal R, Pandey G, Sharma P. et al. Biochemistry of Microbial Degradation of Hexachlorocyclohexane and Prospects for Bioremediation. Microbiology and Molecular Biology Reviews. 2010;74:58-80
16. Sharma P, Raina V, Kumari R. et al. Haloalkane dehalogenase LinB is responsible for beta- and delta-hexachlorocyclohexane transformation in Sphingobium indicum B90A. Appl Environ Microbiol. 2006;72:5720-5727
17. Dogra C, Raina V, Pal R. et al. Organization of lin genes and IS6100 among different strains of hexachlorocyclohexane-degrading Sphingomonas paucimobilis: evidence for horizontal gene transfer. J Bacteriol. 2004;186:2225-2235
18. Concha-Gran E, Turnes-Carou MI, Muniategui-Lorenzo S. et al. Evaluation of HCH isomers and metabolites in soils, leachates, river water and sediments of a highly contaminated area. Chemosphere. 2006;64:588-595
19. Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821-829
20. Promponas V, Enright AJ, Tsoka S. et al. CAST: an iterative algorithm for the complexity analysis of sequence tracts. Complexity analysis of sequence tracts. Bioinformatics. 2000;16:915-922
21. Nalbantoglu OU, Way SF, Hinrichs SH. et al. RAIphy: phylogenetic classification of metagenomics samples using iterative refinement of relative abundance index profiles. BMC Bioinformatics. 2011;12:41
22. Altschul SF, Gish W, Miller W. et al. Basic local alignment search tool. J Mol Biol. 1990;215:403-410
23. Huson DH, Auch AF, Qi J. et al. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377-386
24. Dupont CL, Rusch DB, Yooseph S. et al. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J. 2012;6:1186-1199
25. R Core Team. 2015 R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. http://www.R-project.org/
26. Noguchi H, Park J, Takagi T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006;34:5623-5630
27. Hyatt D, LoCascio PF, Hauser LJ. et al. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics. 2012;28:2223-2230
28. Tatusov RL, Natale DA, Garkavtscev IV. et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22-28
29. Punta M, Coggill PC, Eberhardt RY. et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290-D301
30. Kanehisa M, Goto S, Kawashima S. et al. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277-D280
31. Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics. 2010;26:715-721
32. Oksanen JF, Blanchet G, Kindt R. et al. Vegan: Community Ecology Package. 2011; R package version 2.0-5.
33. Ye Y, Doak TG. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput Biol. 2009;5:e1000465
34. Abhilash PC, Singh N. Multiple residue extraction for organochlorine pesticides in medicinal plants. Bull Environ Contam Toxicol. 2008;81:604-607
35. Anjum R, Malik A. Evaluation of mutagenicity of wastewater in the vicinity of pesticide industry. Environ Toxicol Pharmacol. 2013;35:284-291
36. Srivastava AK, Trivedi P, Srivastava MK. et al. Monitoring of pesticide residues in market basket samples of vegetable from Lucknow City, India: QuEChERS method. Environ Monit Assess. 2011;176:465-472
37. Sievers S, Friesel P. Soil contamination patterns of chlorinated organic compounds: looking for the source. Chemosphere. 1989;19:691-698
38. Chatterjee DK, Kellogg ST, Watkins DR. et al. Molecular Biology, Pathogenicity, and Ecology of Bacterial Plasmids. In: (ed.) Levy SB, Clowes RC, Koenig EL. Plasmids in the Biodegradation of Chlorinated Aromatic Compounds. US: Springer US. 1981:519-528
39. Lal R, Dogra C, Malhotra S. et al. Diversity, distribution and divergence of lin genes in hexachlorocyclohexane-degrading sphingomonads. Trends Biotechnol. 2006;24:121-130
40. Nagata Y, Ohtomo R, Miyauchi K. et al. Cloning and sequencing of a 2,5-dichloro-2,5-cyclohexadiene-1,4-diol dehydrogenase gene involved in the degradation of gamma-hexachlorocyclohexane in Pseudomonas paucimobilis. J Bacteriol. 1994;176:3117-3125
41. Pukatzki S, Ma AT, Sturtevant D. et al. Identification of a conserved bacterial protein secretion system in Vibrio cholera using the Dictyostelium host model system. Proc Natl Acad Sci U S A. 2006;103:1528-1533
42. Schell MA. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993;47:597-626
43. Cornelis GR, Agrain C, Sorg I. Length control of extended protein structures in bacteria and bacteriophages. Curr Opin Microbiol. 2006;9:201-206
44. Boyer F, Fichant G, Berthod J. et al. Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: What can be learned from available microbial genomic resources? BMC Genomics. 2009;10:104
45. Zheng J, Leung KY. Dissection of a type VI secretion system in Edwardsiella tarda. Mol Microbiol. 2007;66:1192-1206
46. Aschtgen MS, Bernard CS, Bentzmann SD. et al. SciN is an outer membrane lipoprotein required for type VI secretion in enteroaggregative Escherichia coli. J Bacteriol. 2008;190:7523-7531
47. Kanamaru S. Structural similarity of tailed phages and pathogenic bacterial secretion systems. Proc Natl Acad Sci U S A. 2009;106:4067-4068
48. Kostakioti M, Newman CL, Thanassi DG. et al. Mechanisms of Protein Export across the Bacterial Outer Membrane. J Bacteriol. 2005;187:4306-4314
49. Howea AC, Janssonc JK, Malfattic SA. et al. Tackling soil diversity with the assembly of large, complex metagenomes. Proc Natl Acad Sci U S A. 2014;111:4904-4909
Author contact
![]() Corresponding author: Molecular Microbiology, Molecular Biology Laboratory, Department of Zoology, University of Delhi, Delhi-110007, India. Tel: (91) 11-27666254, Fax (91) 11-27666254 Email: ruplalcom
Corresponding author: Molecular Microbiology, Molecular Biology Laboratory, Department of Zoology, University of Delhi, Delhi-110007, India. Tel: (91) 11-27666254, Fax (91) 11-27666254 Email: ruplalcom
Citation styles
APA
Negi, V., Lal, R. (2017). Metagenomic Analysis of a Complex Community Present in Pond Sediment. Journal of Genomics, 5, 36-47. https://doi.org/10.7150/jgen.16685.
ACS
Negi, V.; Lal, R. Metagenomic Analysis of a Complex Community Present in Pond Sediment. J. Genomics 2017, 5, 36-47. DOI: 10.7150/jgen.16685.
NLM
Negi V, Lal R. Metagenomic Analysis of a Complex Community Present in Pond Sediment. J Genomics 2017; 5:36-47. doi:10.7150/jgen.16685. https://www.jgenomics.com/v05p0036.htm
CSE
Negi V, Lal R. 2017. Metagenomic Analysis of a Complex Community Present in Pond Sediment. J Genomics. 5:36-47.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.