ISSN: 1839-9940

 Global reach, higher impact
Global reach, higher impactJ Genomics 2017; 5:71-74. doi:10.7150/jgen.20915 This volume Cite
Short Research Paper
Complete Genome Sequence and Comparative Genomics of a Streptococcus pyogenes emm3 Strain M3-b isolated from a Japanese Patient with Streptococcal Toxic Shock Syndrome
Kohei Ogura1, Shinya Watanabe1, Teruo Kirikae2, Tohru Miyoshi-Akiyama1 ![]()
1. Pathogenic Microbe Laboratory
2. Department of Infectious Diseases, Research Institute, National Center for Global Health and Medicine, 1-21-1 Toyama, Shinjuku-ku, Tokyo 162-8655, Japan
Received 2017-5-8; Accepted 2017-6-21; Published 2017-7-1
Abstract

Epidemiologic typing of Streptococcus pyogenes (GAS) is frequently based on the genotype of the emm gene, which encodes M/Emm protein. In this study, the complete genome sequence of GAS emm3 strain M3-b, isolated from a patient with streptococcal toxic shock syndrome (STSS), was determined. This strain exhibited 99% identity with other complete genome sequences of emm3 strains MGAS315, SSI-1, and STAB902. The complete genomes of five additional strains isolated from Japanese patients with and without STSS were also sequences. Maximum-likelihood phylogenetic analysis showed that strains M3-b, M3-e, and SSI-1, all which were isolated from STSS patients, were relatively close.
Keywords: Streptococcus pyogenes, complete genome sequence, streptococcal toxic shock syndrome.
Introduction
Lancefield group A Streptococcus pyogenes (GAS), subtyped by the emm gene encoding M protein, is a non-motile, non-spore forming, beta-hemolytic Gram-positive bacterium belonging to family Streptococcaceae, order Lactobacillales, class Bacilli. GAS causes a wide variety of infectious diseases, which range in severity from relatively benign to life threatening. GAS harboring emm1 has been reported in patients with streptococcal toxic shock syndrome (STSS), a life-threatening GAS infection [1-3]. Extensive studies of the evolution of a highly virulent clone of GAS emm1 have shown the importance of multiple horizontal gene transfer events. From 2010 to 2012, the predominant GAS genotype isolated from patients with STSS in Japan was found to be emm1, followed by emm89, emm12, emm28, emm3, and emm90 in order of their prevalence [4]. Although less is known about the evolutionary and genetic events occurring in emm3 than in emm1 isolates associated with STSS, GAS emm3 strains are responsible for both STSS and pharyngitis [5-7].
Sequencing of 95 GAS emm3 genomes isolated from patients in the province of Ontario, Canada, resulted in the identification of 280 biallelic single nucleotide polymorphisms (SNPs) [7]. The complete genomes of three GAS emm3 strains have been sequenced. Strains MGAS315 and SSI-1 were isolated from STSS patients in 1986-1990 and 1994, whereas strain STAB902 was from a non-invasive superficial cutaneous infection in 2011 [10-12]. This study reports the complete genome sequence of GAS emm3 strain M3-b, isolated in 1994 from a patient with STSS in Japan. The draft genome sequences of an additional five strains isolated in Japan were also determined.
Genome Announcement
Two types of sequencing platform were utilized, 454 and Illumina. An 8-kb paired-end library was generated for 454 sequencing. Sequencing with the 454 platform was performed to a 38.7-fold depth of coverage and was used to assemble an initial draft scaffold of the M3-b genome. The pair-end library of the GAS M3-b genome was prepared and sequenced using GS junior according to the manufacturer's instructions (Roche). This generated 445,292 reads and 73,214,557 bp of sequence (38.7-fold coverage), which were assembled into contigs and scaffolds. Gaps were filled by conventional Sanger sequencing of the PCR fragments based on brute force PCR among the contigs and scaffolds. The assemblies were verified by mapping of reads generated from Miseq (Illumina).
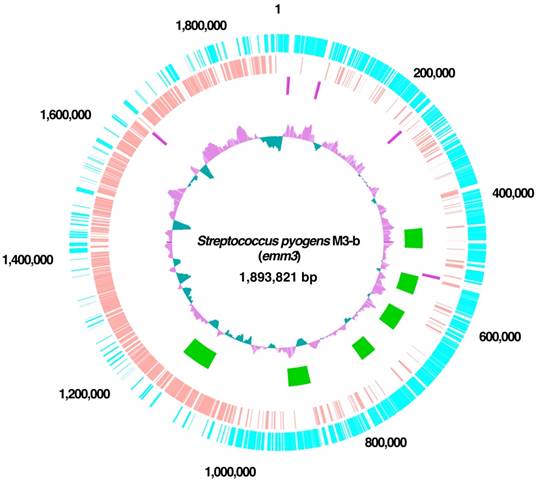
The M3-b genome was found to consist of a single circular 1,893,821 bp chromosome, with an average GC content of 38.54% (Table 1 and Figure 1). The complete nucleotide sequence of the chromosome of GAS M3-b has been deposited in DDBJ under accession number AP014596. Its genome was annotated by using Glimmer 3.02 [8] to extract its primary coding sequence (CDS), with initial functional assignment and manual correction performed by genome editing commercial software (in silico molecular cloning; in silico biology, inc). The annotated chromosome contained 1926 protein-encoding genes and 58 tRNA-encoding genes for all amino acids (Table 2). PHAST showed that the chromosome harbored six prophage-like elements, at nucleotides 443879-486292, 545445-585108, 624934-669255, 722262-761492, 877665-924352, and 1098269-1160870 [9]. Each of these six prophage-like genes was highly conserved, with >80% sequence identity relative to the emm3 strains MGAS315, SSI-1, and STAB902. CRISPRFinder [13] showed that these four strains did not contain functional or functionally active clustered, regularly interspaced short palindromic repeats (CRISPRs), suggesting that these emm3 strains were prone to infection by phages.
Project information
| Property | Term |
|---|---|
| Finishing quality | Level 6, finished |
| Libraries used | 8 kb paired-end library |
| Sequencing platforms | 454 platform |
| Fold coverage | 38.7- |
| Assemblers | Newbler 2.7 |
| Gene calling method | Glimmer 3.02 |
| Locus tag | M3_b |
| Genbank ID | AP014596 |
| GenBank date of release | February 25, 2016 |
| GOLD ID | Gs0118386 |
| BIOPROJECT | PRJDB1740 |
| Source material identifier | M3-b |
| Project relevance | Human pathogen |
Circular representation of the genome of S. pyogenes strain M3-b. Circle 1 (outermost circle) indicates the distances from the putative origin of replication. Circles 2 and 3 show annotated CDS encoded by the forward (light blue) and reverse (pink) chromosomal strands, respectively. Circle 4 shows the rrs operons. Circle 5 shows prophages (green). Circle 6 (innermost circle) shows the G+C content with more and less than average (0.40) in purple and green, respectively.
Genome statistics
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 1,893,821 | 100 |
| DNA coding (bp) | 1,633,134 | 86.2 |
| DNA G+C (bp) | 729950 | 38.5 |
| DNA scaffolds | 1 | |
| Total genes | 2002 | 100 |
| Protein coding genes | 1926 | 96.2 |
| RNA genes | 76 | 3.8 |
| Pseudo genes | 0 | - |
| Genes in internal clusters | 0 | - |
| Genes with predicted function | 1498 | 74.8 |
| Genes assigned to COGs | 1610 | 80.4 |
| Genes with Pfam domains | 1571 | 78.5 |
| Genes with signal peptides | 96 | 4.8 |
| Genes with transmembrane helices | 416 | 20.8 |
| CRISPR repeats | 0 | - |
Two other emm3 strains, M3-a and M3-e, were isolated from STSS patients in Japan in 1994 and 1993, respectively, whereas three additional emm3 strains, M3-1, M3-3, and M3-4, were isolated from pharyngitis patients in Japan in 1985, 1994, and 1984, respectively. These five additional emm3 strains (M3-1, M3-3, M3-4, M3-a and M3-e) were newly sequenced using a MiSeq system with Nextera XT library kits (Illumina). Approximately one million 301 bp x 2 pair-end reads were obtained. After trimming based on base quality (quality score limit = 0.05, removing reads with more than two ambiguous nucleotides and those <15 bp in length), the reads were de novo assembled to construct contigs without annotation using a commercial software program, CLC genomics workbench (CLC bio). The contigs were used for further analyses. The raw reads data have been registered with DDBJ as accession number DRA003035.
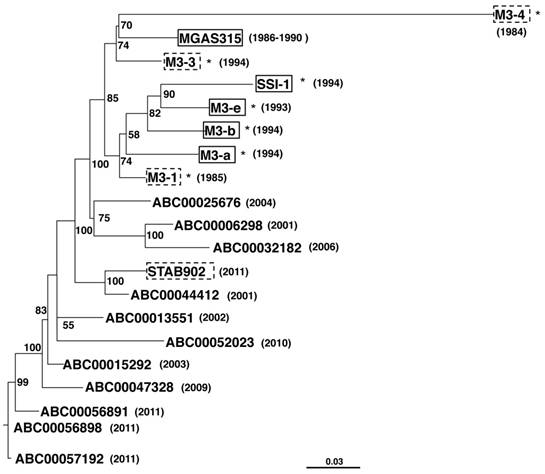
All five strains possessed phages encoding the speA, sdn, and DNase genes, with four of these strains, all except strain M3-4, harboring speL, which encodes streptococcal pyrogenic exotoxin L. Maximum-likelihood phylogenetic analysis by of core genomes PhyML 3.0 [14] showed that the strains M3-a, M3-b, M3-e, and SSI-1, all of which were isolated from patients with STSS, were closely related (Figure 2). However, strain MGAS315, which was also isolated from a patient with STSS, was more closely related to strains M3-3 and M3-4, which were isolated from non-STSS patients. These results indicated that emm3 isolates from patients with and without STSS were indistinguishable.
Maximum-likelihood tree of GAS emm3 strains isolated in Japan. The phylogenetic tree was prepared using concatenated SNPs. The tree model was related to HKY models using jModel Test [15] and calculated with PhyML [14] with 100 bootstrappings, showing the indicated values for each branch. Trees were visualized using FigTree. Asterisks represent the strains isolated in Japan. Strains derived from STSS and non-STSS patients are indicated in solid-line and dashed line boxes, respectively. Years of isolation are indicated in parentheses.
Acknowledgements
The authors thank Mrs. Komiya and Mrs. Sakurai for excellent work in the genome analysis. This work was partly supported by a Grant for Research on Emerging and Reemerging Infectious Diseases (H22 Shinkouh-013), by JSPS KAKENHI Grant Numbers 24390109 (TMA), 25860330 (SW), 15H05654 (SW), 15K18977 (KO), and 15H04734 (TMA), and by a Grant for International Health Research (26A-103) from the Ministry of Health, Labour and Welfare of Japan (TK). This research is partially supported by the Research Program on Emerging and Re-emerging Infectious Diseases from Japan Agency for Medical Research and development, AMED.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Miyoshi-Akiyama T, Zhao J, Kikuchi K, Kato H, Suzuki R, Endoh M, Uchiyama T. Quantitative and qualitative comparison of virulence traits, including murine lethality, among different M types of group A streptococci. J Infect Dis. 2003;187:1876-87
2. Ekelund K, Darenberg J, Norrby-Teglund A, Hoffmann S, Bang D, Skinhøj P, Konradsen HB. Variations in emm type among group A streptococcal isolates causing invasive or noninvasive infections in a nationwide study. J Clin Microbiol. 2005;43:3101-9
3. Shea PR, Ewbank AL, Gonzalez-Lugo JH, Martagon-Rosado AJ, Martinez-Gutierrez JC, Rehman HA, Serrano-Gonzalez M, Fittipaldi N, Beres SB, Flores AR, Low DE, Willey BM, Musser JM. Group A Streptococcus emm gene types in pharyngeal isolates, Ontario, Canada, 2002-2010. Emerg Infect Dis. 2011;17:2010-7
4. Ikebe T, Tominaga K, Shima T, Okuno R, Kubota H, Ogata K, Chiba K, Katsukawa C, Ohya H, Tada Y, Okabe N, Watanabe H, Ogawa M, Ohnishi M. Increased prevalence of group A streptococcus isolates in streptococcal toxic shock syndrome cases in Japan from 2010 to 2012. Epidemiol Infect. 2015;143:864-72
5. O'Loughlin RE, Roberson A, Cieslak PR, Lynfield R, Gershman K, Craig A, Albanese BA, Farley MM, Barrett NL, Spina NL, Beall B, Harrison LH, Reingold A, Van Beneden C. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45:853-62
6. Olsen RJ, Shelburne SA, Musser JM. Molecular mechanisms underlying group A streptococcal pathogenesis. Cell Microbiol. 2009;11:1-12
7. Beres SB, Carroll RK, Shea PR, Sitkiewicz I, Martinez-Gutierrez JC, Low DE, McGeer A, Willey BM, Green K, Tyrrell GJ, Goldman TD, Feldgarden M, Birren BW, Fofanov Y, Boos J, Wheaton WD, Honisch C, Musser JM. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc Natl Acad Sci U S A. 2010;107:4371-6
8. Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636-41
9. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39(Web Server issue):W347-52
10. Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu M-Y, Smoot JC, Porcella SF, Parkins LD, Campbell DS, Smith TM, McCormick JK, Leung DYM, Schlievert PM, Musser JM. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci U S A. 2002;99:10078-83
11. Nakagawa I, Kurokawa K, Yamashita A, Nakata M, Tomiyasu Y, Okahashi N, Kawabata S, Yamazaki K, Shiba T, Yasunaga T, Hayashi H, Hattori M, Hamada S. Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Res. 2003;13:1042-55
12. Soriano N, Vincent P, Moullec S, Meygret A, Lagente V, Kayal S, Faili A. Closed genome sequence of noninvasive streptococcus pyogenes M/emm3 strain STAB902. Genome Announc. 2014;2:e00792-14
13. Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35(Web Server issue):W52-7
14. Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307-21
15. Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772
Author contact
![]() Corresponding author: Tohru Miyoshi-Akiyama, Pathogenic Microbe Laboratory, Department of Infectious Diseases, Research Institute, National Center for Global Health and Medicine, 1-21-1 Toyama, Shinjuku-ku, Tokyo 162-8655, Japan. Phone: +81-3-3202-7181, ext. 2903. Fax: +81-3-3202-7364. E-mail: takiyamncgm.go.jp.
Corresponding author: Tohru Miyoshi-Akiyama, Pathogenic Microbe Laboratory, Department of Infectious Diseases, Research Institute, National Center for Global Health and Medicine, 1-21-1 Toyama, Shinjuku-ku, Tokyo 162-8655, Japan. Phone: +81-3-3202-7181, ext. 2903. Fax: +81-3-3202-7364. E-mail: takiyamncgm.go.jp.